|


Prusiner傳記:
Prusiner,1942年5月28日於美國中西部出生。父親,Lawrence是美國海軍,因工作的關係,在他的童年早期父親是缺席的。母親Miriam則帶著他跟隨其父親工作的調動,遷移在Des
Moines, Iowa, Cincinnati和 Ohio之間。在就讀Walnut Hills高中期間,為了更能無所限的書寫科學報告,他有計畫的研讀了五年的拉丁文。而後,進入Pennsylvania大學主修化學。1963年夏天以『低體溫法於部分外科手術的應用』開始了他醫學研究之路,之後的醫學養成教育裡,關於棕色脂肪(brown
fat)的新陳代謝研究,則佔據了他大部分的時間。 1969年到1972年,他在美國國家保健局(NIH)的Earl Stadtman's實驗室工作,從事關於E.coli的麩胺酸脢(Glutaminases)的研究,Prusiner自言,在他的科學教育中,這三年的研究訓練對他而言是非常重要的。(1)
※ 從一個諾貝爾獎得主的成長過程,可以發現Prusiner從小就很自主性的發展自己的興趣,期充分的配合所學而有所發揮。
★ 進入下列網站,可瀏覽Prusiner研究室的一些即時影像——
http://www.nobelchannel.com/prizes/przw_vid.sps?id=160&category=Medicine&year=1997
前言:
分子生物學的中心法則(central
dogma)是從DNA製造RNA,然後RNA轉譯蛋白質,蛋白質發揮各種結構性功能,形成種種生命現象。這個中心法則除了十餘年前核酸反轉錄酶(reverse
transcriptase)被發現,確定RNA可以被反轉錄成為DNA後,很少有人能夠提出修改中心法則的假說。(14)
1997 年諾貝爾生理醫學獎頒發給美國加州大學舊金山分校的史坦利‧布魯希納 (Stanley Prusiner) 教授。這項殊榮是肯定布魯希納教授在研究引起人類腦神經退化而成痴呆的古茲菲德-雅各氏病(Creutzfeldt-Jakob
disease, CJD) 病原體的貢獻。其實與 CJD 相類似的疾病還有人類的古魯症 (Kuru)、GSS 氏病(Gerstmann-Straussler-Scheinker
disease)、山羊和綿羊的羊搔癢病 (scraple) 以及牛群中的狂牛病 (mad cow disease) 。它們都是由類似病原體所引起腦神經退化,而產生的疾病。
自 1987 年以來,布魯希納是繼利根川進 (Susumu Tonegawa) 之後單人獲得諾貝爾醫學獎,過去五十年也只有十人享有單人獲得醫學獎的殊榮,更顯示出布魯希納卓越的貢獻。然而對於布魯希納獲獎,《科學》雜誌評論指出,關於
CJD 病原體的研究還尚未徹底,是否尚有其他因子或未知病毒的參與至今還無法完全排除,諾貝爾獎金委員會的答辯認為這種新病原體許多相關問題確實尚未完全釐清,但是對布魯希納發現
prion (proteinaceous infectious particle,具有感染力的蛋白顆粒,本刊之前譯為「普恩蛋白」) 的存在以及闡明
prion 在CJD的致病過程所扮演的角色是值得肯定而獲獎的,諾貝爾獎金委員會甚至預測在 prion 研究的領域中,還會再誕生一些諾貝爾得主。(3)
有關普恩蛋白(Prion protein, PrP)的研究可說是一日千里,主要是因為它的爭議性,在研究與了解越多之後發現,普恩蛋白似乎與一些慢性神經性的疾病有某些關聯性……。(15)
動機:
1972年,Prusiner在加州大學舊金山分校醫學院擔任神經科住院大夫時,有位病人被庫賈氏病(CJD, Creutzfeldt-Jakob
disease)奪走性命,使他對此病發生了濃厚的興趣。為了獲得更多的研究資源,他不惜巧立名目,向衛生研究所申請研究羊搔症(Scrapie;是一種傳染性海綿樣腦病變)的大額研究經費。繼又到瑞典鑽研病毒學,學成回國後,借用英國在六十年代發展出來的技術,設計出一套評估方法,採用潛伏期僅老鼠一半的倉鼠作為實驗動物,大量而快速的獲得實驗數據結果,全心全意追求諾貝爾獎。(1)
※ Prusiner以實際上人類所面對的疾病,定為研究素材,利用經濟產物問題的相關領域,一起評估其相類似的目標物質—prion。
※ 加上其篩選倉鼠作為實驗的材料,使得實驗的步調以及效率提高。
得獎經歷:
1982年,Prusiner在科學雜誌刊出『引起羊搔症的傳染性新蛋白質粒子』的論文,並為其命名為普粒子(Prion),將Prion定義為:一種完全不含核酸的傳染性蛋白質或少量核酸外圍包有緊密的蛋白質外殼;這動作在科學界曾引起強烈的質疑與爭論,因為相同研究領域的科學家們認為,Prusiner的證據太過牽強且並不了解它們的生化性質,論文結論中閃爍其詞,搶先命名,操之過急;Prusiner虛張聲勢的冒失行徑,事實上是一種賭博,只要他的研究有收穫就勝算在握。在一片的伐噠聲中,Prusiner仍然契而不捨越做越起勁。
同年十二月,Prusiner和他的同事就宣稱,已經從感染羊搔症的倉鼠大腦中,精練出一種似乎與感染有關的蛋白質-組織感染程度越嚴重,他們取得的蛋白質就越多。次年,他們報導說,他們精練的蛋白質(現稱為普粒子蛋白)具有「桿狀顆粒」的外型,並成功的用剛果紅為他們染色,染色後就很像澱粉樣蛋白。每個桿狀物含有約一千個普粒子蛋白,他們的發現提升了傳染性,導致退化的神經性疾病中發現的澱粉樣蛋白,係由普粒子組成之可能性。(1)
這些疾病,特別是其中的傳播性退化性中樞神經系統疾病均可在病理解剖中看到細胞內澱粉樣變化。這種病理變化與對prion型態學研究的結果一致。以前,人們一直認為電份樣變化是疾病過程中形成的一種病理性變化。現在,由於對prion研究的深入,使人們不能不聯想到病理學上的澱粉樣變化,實質上可能就是由PrP組成的晶體結構。(18)
續又解開了普粒子蛋白的化學密碼,利用此種蛋白質的基因排列順序,在基因庫裡找到對應的DNA排列順序。更令人吃驚的發現是,它們也存在正常倉鼠及正常人的細胞之中。Prusiner和他的同事接著證實了,正常和患病兩種形式的普粒子蛋白有完全相同的基因密碼。並發現他們只有一處不同的差異而這點非常的重要:正常的普粒子蛋白很容易被一種消化蛋白質的酵素所分解,但病態的普粒子蛋白完全無法消化。這代表大腦會製造正常的普粒子蛋白,但正常的普粒子蛋白有時會變成病態的形式。Prusiner相信這變化是Prion所引起的:不正常的普粒子蛋白的潛伏,以某種方式使正常的普粒子蛋白變成致病的形式,如果普粒子蛋白改變形式,細胞可能就停止生產正常的形式,而開始製造病態的形式。這也就說明了為什麼致病因子不需要複製核酸,數量還是不斷的增加。
Prusiner是第二位因海綿樣腦病變而得到諾貝爾獎的學者,第一位是加德塞克(Carleton
Gajdusek)早在1976年10月因其對新幾內亞所發生的克魯症(Kuru)也是一種傳染性海綿樣腦病變)之研究而得獎。Prusiner的發現,對於傳染性海綿樣腦病變的生物機制提供了重要的透析。並且對藥物的研發和新的醫療策略建立了基礎。這是人類首次發現蛋白質能夠致命,會不會幾年後又有第三位科學家因為此種腦病變而又得到諾貝爾獎呢?這是我們所期待而又拭目以待的事。(1)
※ 因此我們可以知道,Prusiner得到諾貝爾獎不是沒有原因的,他必須打破傳統觀念,科學家之間的明爭暗鬥,以及搶先發表論文,且須提出強而有力的實驗證據。
新病原體prion:
自從1997年的諾貝爾生理醫學獎,頒發給了美國加州大學舊金山分校的史坦利‧布魯希納(Stanley Prusiner)教授之後,大家就對這種新病原體非常的好奇。布魯希納教授研究的是什麼呢?他研究的是一種能夠引起人類腦神經退化、形成痴呆的庫茲菲德-雅各氏症(Creutzfeldt-Jakob
disease),簡稱庫雅氏症,英文簡稱為CJD,這種疾病的病原體,在病人體內,他發現了一種具有感染力的蛋白質顆粒,稱做普恩蛋白質prion
(proteinaceous infectious particle)。
究竟什麼是普恩蛋白質呢?早在1913年左右,德國有一個修道院,有位女僕,因為得了一種未知名的疾病,在發病後只有短短的三個月就死亡了,為她治病的庫茲菲德醫師記錄了她所有的病程,發現這位女僕的腦部完全沒有發炎,但是卻嚴重的受損;後來另外又有一位醫師,名叫雅各,他也發現了一位患者,情形與那位女僕幾乎完全相同。因為這二位醫師的發現,這一種未知的疾病,就被命為庫茲菲德-雅各氏症(簡稱庫雅氏症,或CJD)。
1950年時,在新幾內亞高地有一個食人的原始部落,稱作富雷部落,爆發了一種未知的疾病,這種疾病在該部落內漫延開來,造成該部落許多婦女及小孩得病而死,當地的人稱此病為枯魯(kuru)。為了揭開這傳染病之謎,1957年有一位美國病毒學專家名叫加德賽克醫師,和另一位名叫吉加斯的醫師,他們兩個人一起進入富雷部落,著手研究該致命的疾病;他們發現病人先會出現運動失調的現象,之後會變成癡呆,然後才死亡;加德賽克與吉加斯在患者死亡後,解剖死者的腦組織時,意外發現死者的腦組織損壞嚴重,呈海綿狀有許多小空洞,他們同樣的發現了一件驚人的事實,患者的腦沒有發炎,但卻嚴重的受損。
在隨後的幾年之中,陸陸續續的又在幾個不同的國家,分別傳出了類似的疾病,不論是在人類、或在動物身上都有許多新的發現;例如在英國的羊搔癢症(Scrapie)、在美國的傳染性貂腦病(TME,transmissible mink encephalopathy),甚至最近由1996年起在英國爆發的瘋牛症;發病時的病徵都與前述情況相似。
不論是使人類或是使動物致病,這一類疾病共同的特徵,都是會引起健康人或動物的感染。一般的傳染性疾病,都會有某種的病原體,而這一類的疾病,卻一直找不到任何的已知的病原體。
Kuru症一般症狀是失去運動性的協調,然後變成癡呆,從1957年以來,共有2600個案例,發病期間是三個月至一年。加德賽克醫師後來曾經以黑猩猩作實驗,證明Kuru是可以傳染的,他推測Kuru病的病原體,是一種未知的慢作用性病毒(slow-acting
virus)。他協助富雷的原住民,杜絕了分食逝世親人屍體腦部的風俗之後,Kuru從此就消失了。加德賽克醫師因此獲得了1976年的諾貝爾醫學獎。但是科學家們也在加德賽克的諾貝爾獎光環之下,一致認為有一種慢病毒,侵入人或動物的體內就躲起來,等到慢慢作用了許久之後,才突然發病。科學家們一直在找這種慢病毒,但是長久以來,這種慢病毒一直是科學之謎。(2)
※ 這種”慢性病毒”就是後來命名的prion。
奇異的病原體:
引起人類和動物疾病的病原體種類很多,按這些病原體結構的複雜成度,以及個體的大小,病原體包括多細胞真核生物的線蟲、吸蟲、絛蟲、真菌,單細胞真核的原生生物、原核生物的細菌,介於細菌和病毒間的立克次體、披衣菌,以及不具有任何細胞結構的病毒、類病毒等。但不論病原體是什麼,都會含有特定的遺傳物質。(表一)
具有細胞結構的,不論是多細胞或單細胞,是真核生物或原核生物,都以DNA為其遺傳物質。病毒則有DNA病毒或RNA病毒,病毒已經簡單到只含蛋白質和核酸了,而且只能利用電子顯微鏡,才能觀察到它們的結構;但是引起CJD的病原,普恩蛋白質,僅僅含有蛋白質而已,並沒有什麼核酸物質,也就是說,所謂的慢病毒並不存在。
蛋白質具有傳染力,並能引起疾病,這實在是一件很不可思議的事!因為不論是DNA病毒或是RNA病毒、類病毒,病原體最少都是以核酸作為其遺傳物質,去繁殖更多的病原體。今天這個使人類或動物致病的奇異致病源,竟然是一種可以自行複製增殖的蛋白質。
基本上,普恩蛋白質是一種體積小、具有傳染力的蛋白質顆粒,對絕大多數用來抑制核酸活動的處理方式,都具有抵抗力。相對地,它也破壞了所謂的生物中心法則:也就是一般的遺傳信息流動都先由DNA做出RNA,再由RNA做出蛋白質。因此,蛋白質要進行複製時,基本上都需要有DNA及RNA的存在,才能進行基因的複製與遺傳。可是,普恩蛋白質沒有DNA也沒有RNA,同樣可以進行複製增殖的反應。
由普恩蛋白質導致的疾病有哪些呢?到目前為止,普恩蛋白質所引起的疾病都是哺乳類動物的專利,包括羊隻的搔癢症、牛隻的瘋牛症(BSE,bovine spongiform encephalopathy)、貓科動物的傳染性貂腦病(TME,transmissible
mink encephalopathy)、鹿及麋鹿等動物的消耗症(Wasting disease)、人類的Kuru、庫雅症、運動失調的GSS症,(Gerstmann-Straussler-Scheinker
syndrome)、及致命性家族失眠症(familial insomnia)。
普恩蛋白質引起的疾病,特徵都是在腦部與中樞神經中出現海綿狀的組織,或是出現很多的空洞,而人類的阿茲海默症似乎也有類似的組織出現,形成海綿狀腦部病變(spongiform encephalopathy)。
比較患病的人或動物大腦皮層切片,這些大腦中的「洞」,形成了我們所說的CJD,症狀包括運動協調性喪失、癡呆症、以及死亡,以及腦部充滿了許多洞,讓腦部像海綿一樣,瘋牛症也是一種傳染性海綿樣腦病變。
庫雅氏症CJD一般症狀為癡呆,然後失去運動的協調,或者兩者順序對調,引起腦神經退化再形成痴呆,與Kuru相似,但卻是一種全球性的疾病,感染途徑通常原因不明,發生率是百萬分之一,但大多發生在六十歲左右的人,其中10%-15%是屬於遺傳性,其餘亦包括醫療手術引起的,如:眼角膜的移植、腦部手術污染、或是注射人類腦下垂體提取的生長激素;屬感染型的有八十個案例。目前已有超過一百個家庭被證實是由於基因突變造成,通常一年,但也有短到一個月、或是長到十年以後才發病的。
運動失調症的GSS (Gerstmann-Straussler-Scheinker
syndrom)病,和致命性的家族失眠症,都是屬於遺傳性的疾病,不過患者大都要等到中老年的時候才會發病。GSS病一般的症狀,是先失去協調然後變成癡呆,在五十多個家族被證實是遺傳性PrP基因突變,發病期間通常為二至六年。而致命家族失眠症的症狀則為無法睡眠,自主神經失調,然後嚴重性失眠而癡呆,在六個家族中被證實為遺傳性的PrP基因突變,發病期間通常將近一年。(2)
(表二)
|
表一
|
|
生物種類或成分
|
病原體名稱
|
引起疾病
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
肝硬化、肝癌
|
|
紅疹、斑診、出
|
|
血性登革熱則會
|
|
休克而死
|
|
|
|
|
|
這些由普恩蛋白質引起的疾病,都會在腦部與中樞神經中出現海綿狀的組織,或是出現很多的空洞,統稱為傳染性海綿樣腦病變。這些疾病的患者都會有運動協調性喪失、癡呆、及死亡的狀況發生。(2)
|
表二
|
|
疾病
|
一般症狀
|
感染途徑
|
疾病分布
|
發病期間
|
|
古魯症
|
失去協調然後痴呆
|
可是經過分食屍腦感染
|
新幾內亞高地1957年來共有2600案件
|
三個月至一年
|
|
古茲菲德‧雅各氏病
|
痴呆然後失去運動協調,或者兩者順序相對調
|
(1)通常原因不明(由其是那些散失性的)
(2)由於基因突變造成的
(3)醫療手術引起
|
(1)百萬分之一的發生率
(2)已超過一百個家族被證實
(3)屬感染型的有八十案例
|
通常一年,但也有一個月或十年以後才發病
|
|
GSS病
|
失去協調然後變成痴呆
|
遺傳性PrP基因突變
|
五十多個家族被證實
|
通常二至六年
|
|
致命家族失眠症
|
無法睡眠自主神經失調然後嚴重性失眠而痴呆
|
遺傳的性PrP基因突變
|
六個家族被證實
|
通常將近一年
|
(3)
以下是一些prion disease的組織切片圖:
|
This is your Brain . . . . . . . this is your Brain on Beef
|
|
CJD - human
|
Kuru -
human
|
|
![[239_SM]](p1997.files/image003.gif)
|
![[235_SM]](p1997.files/image004.gif)
|
|
BSE - (cow)
|
Scrapie -
(sheep)
|
|
![[KUBO8_SM]](p1997.files/image005.gif)
|
![[scrapie1_SM]](p1997.files/image006.gif)
|
(12)
Prion致病的科學證據:
1982年,布魯希納醫師在其實驗室中,純化出引起CJD病的病原體,並且證明該病原體只有含蛋白質,而不含任何的核酸成分,布魯希納醫師將這新發現、特異的病原體命名為普恩蛋白質,簡稱為PrP。
PrP在結構上有兩種型式,生化分析也發現突變的PrP比正常的PrP更不容易被蛋白酵素所分解,顯示出這兩種蛋白質在型式上的差異。為了區別這兩種不同的PrP,布魯西納就將正常的PrP稱作PrPC,C代表細胞,致病的PrP稱作PrPSC,SC代表羊搔癢病。正常的PrPC和致病突變的PrPSC在結構上不同,正常的PrPC在體內數量相當少,通常不致病。
病人PrP基因突變的發現,支持了兩種型式PrP的假說,突變的PrP是可致病的。一般PrPC轉換成PrPSC的機率並不高,但若PrPC的基因發生突變,則會大幅提高此轉換機率。
為了證明這個假說,布魯希納的實驗室進行了基因轉殖的實驗,將突變的PrP基因種入小鼠的染色體中,讓突變的PrP可以大量產生,結果這些基因轉殖小鼠最後發病了!
PrP可以作為一種病原體也因此被證實了!若將發病轉殖小鼠的腦萃取液打入另外一隻未發過病的轉殖小鼠,則會更快促使牠的發病及死亡,這實驗的結果,更加證實了這個結論。
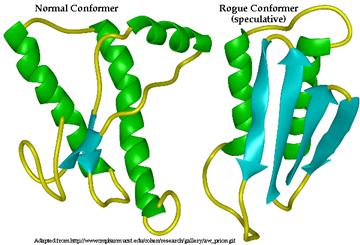
經由氨基酸的定序分析測定,PrPC與PrPSC的胺基酸鏈雖然相同,但以分子模擬,比較PrPC與PrPSC的蛋白質二級結構(secondary
structure),發現PrPC的主要結構是α-螺旋,正常PrPC的胺基酸鏈會折疊成四個α-螺旋。幾乎沒有β-片狀摺疊的結構;但改變後的PrPSC,卻以β-片狀摺疊結構為主,因為突變的胺基酸使PrP無法形成穩定的α-螺旋狀結構,而轉化成致病性的PrPSC,而且PrPSC的溶解度極低。
以前曾有許多學者致力於普恩蛋白質的研究,想以實驗證明PrP就是羊搔癢病、瘋牛病、及CJD等的病原體,以證明PrP致病的特異性。
經由實驗的證實,布魯希納醫師就提出了假說,認為該病原體變形的主要原因有二:一是病原體的基因發生突變;二是基因沒有改變,但是所造出來的蛋白質自己變形了。在1997年,布魯希納醫師因為其所致力研究的普恩蛋白質,而獲得了諾貝爾獎。他是一個很具爭議性的人物。1997年10月的「科學」雜誌,介紹專文中提到:他20年來致力研究庫賈氏病的致病原因,發現感染性蛋白粒子,功不可沒,但某些學者認為這個獎對他是給早了一點。
由於普恩蛋白質引起的疾病,乃在於PrP結構型式的轉變,若能研發出某種藥物,阻止這種轉變,應該就就可以治好這類的疾病。但有些學者推測,會致病的因素可能是『失去了PrPC』而不是『出現了PrPSC』。
普恩蛋白質在體內的功能,極可能與小腦皮層中浦金埃(Purkinje)細胞的長時間存活有關。也有學者認為帕金森氏症的患者會發病,可能或多或少也是因為失去了PrPC;倘若往後能研發出防止失去PrPC結構的藥物,或是調節普恩蛋白質與浦金埃細胞之間的關係,或許可免除與腦神經相關的疾病發生。因此,學者從一未知疾病中找出PrP之後,同時打開了蛋白質誘發蛋白質形式改變的研究之旅。相對地,也可能讓腦部疾病患者、或相關疾病患者得到更好的醫療方法。
至於PrPSC如何誘發正常的PrPC轉變結構?目前有一種結晶的理論,PrPSC進入神經細胞後,就取代原來的PrPC成為晶種,使得以後所製造出的蛋白質結構,會仿照PrPSC的形式,使細胞將充滿不正常的蛋白質。
所以PrPSC 有下列的特徵:(1)它來自正常的蛋白質,卻不具有正常的功能。(2)它與正常蛋白質之間僅有三度空間結構上的差異,卻能引起嚴重的疾病。(3)它能逃避蛋白質水解脢的分解。(4)它能導致正常蛋白質的不正常化。
最新的實驗證據顯示,被感染的普恩蛋白質會摺疊成PrPSC型式,當動物吃入感染的普恩蛋白質,並進入腦部後,PrPSC就像鑄模一樣,改變原正常的普恩蛋白質結構,這感染的過程,發生在正常普恩蛋白質打開摺疊(unfolding)
時。一般也認為,TSE的成因,一部份是由於一種熱休克蛋白(GroEL)無法即時修正普恩蛋白質的錯誤摺疊所致。普恩蛋白質的感染途徑有二,先天遺傳或後天感染。(2)
※ 雖然我們對prion詳細確實的致病因子尚不明瞭,但已找到prion研究的方法,期待未來得到更多prion的background,以得到更完整的定義prion。
PrP 能複製嗎?
蛋白質能複製嗎?生命科學的中心要旨告訴我們:蛋白質的產生是依靠核酸上訊息的藍本,只有核酸具備複製能力,蛋白質無法自形模板加以大量複製。如果 PrP 只含蛋白,如何解釋它進入動物體腦細胞後可以偵測到大量的 PrP 呢?布魯希納實驗室決定找尋 PrP 的基因。 透過與加州理工學院胡德教授
(Leory Hood) 和蘇黎士大學魏斯曼(Charles Weissmann ) 實驗室的合作,他們首先將 PrP 其中十五個胺基酸序列決定出來,然後利用這個胺基酸序列反推核酸序列,並且作成探針尋找
PrP 的相對基因,原來這個基因就在倉鼠的染色體上,之後許多不同實驗室也證明小鼠、人類和許多哺乳類動物都具有 PrP 基因。這些發現不免引人懷疑
PrP 不是引起疾病的元凶?還是 PrP 會有兩種形式:正常的不致病,異常的才會致病? 為了確認 PrP 是否就是病原體,布魯希納實驗室利用基因工程生產的
PrP 打入實驗動物的腦中,結果倉鼠並沒有發病。這個失敗的實驗,讓他們轉移研究方向去分析那些具有遺傳性 GSS 病的 PrP 基因,結果發現病人的
PrP 基因確實有單一核酸的突變。其他的實驗室分析 GSS 病人的PrP 基因也都發現有突變,而這些突變的 PrP 基因會遺傳下來。(3)
兩種形式的 PrP :
病人 PrP 基因突變的發現支持了兩種形式 PrP 的假說,突變的 PrP 是可致病的。為了證明這個假說,布魯希納實驗室進行了基因轉殖的實驗,將突變的
PrP 基因種入小鼠的染色體,讓突變的 PrP 大量產生,結果這些轉殖小鼠最後發病了!PrP 是種病原體被證明了!若將發病轉殖小鼠的腦萃取液打入另外未發病的轉殖小鼠,更快促使他們發病死亡的實驗結果,更加證實這個結論(下圖)。
|

|
|
起先是健康的轉殖小鼠,再產生大量突變PrP之後病發死亡。
|
會製造少量病變PrP的轉殖小鼠在接受粹取液後,此轉殖小鼠也會發病。
|
另一隻會製造少量突變PrP的轉殖小鼠再接受粹取液後,也發病了。
|
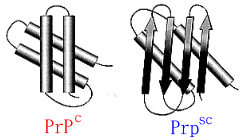
經過分子模擬的分析,致病的 PrP 和正常的 PrP 結構上有所不同,正常的PrP形成四個α-螺旋狀結構(下圖左)而突變的 PrP則為β-片狀結構(下圖右)。
|
|
|
正常的PrPC通常是不致病的,其氨基酸折疊成四次α-螺璇狀(左圖,四個桶狀示意圖),一旦變成致病性PrP (PrPSC),其結構則變成β-片狀結構(右圖,四個上下箭頭的示意圖)。
|
而突變的PrP會比較趨向於摺疊成β-片狀結構,是因為突變的胺基酸使PrP無法形成穩定的α-螺旋狀結構。一些生化的分析,也發現變異的PrP比正常PrP不易被蛋白分解,顯示兩種蛋白的差異性。為了區辨這兩種不同的 PrP,布魯希納就將正常的PrP稱作PrPc(c
代表細胞),致病的PrP稱作PrPsc(sc 代表羊搔癢病)。(3)
兩種形式的PrP:(左為PrPc,右為PrPsc)
(22)
致病 PrP 如何增加?
|
圖三
|
|

|
PrP 是造成 GSS 病的主因,但一些散發性傳染的 CJD 又如何解釋呢?因為患者都擁有正常的 PrP 基因!以 PrP 基因剔除(gene
knockout) 的小鼠進行的實驗提供了一些說明:小鼠在發育過程顯然不需要PrPc,而將PrPsc 打入這些小鼠的腦中,這些小鼠不會發病,也找不到大量的PrPsc,顯然
PrPc 是使PrPsc 增加的藍本,致病的 PrP 可以將正常的 PrP 誘發成致病性的 PrP (圖三)。 誘發的假說是如何建立的?布魯希納曾經嘗試把倉鼠的PrPsc
打入小鼠腦中,都無法造成小鼠發病,但將倉鼠的PrPsc 打入含有倉鼠 PrP 基因的轉殖小鼠,這些小鼠則會產生大量PrPsc,並且發病。這實驗除了提供誘發假說的證據,同時也說明了異種間
PrP 不容易產生誘發,因為倉鼠和小鼠的 PrP 在 254 個胺基酸中有 16 個不同。 誘發的機制目前還不十分清楚,一些轉殖小鼠的實驗結果說明可能需要細胞內伴護子
(chaperone) 的幫忙。若將 CJD、GSS 等病人的腦抽取液注入含有人類全長 PrP 基因的轉殖小鼠和含有人鼠混合 PrP 基因的轉殖小鼠,結果後者發病率較高且較早,顯示也許是人鼠混合
PrPc 比人類PrPc更容易被細胞內未知的伴護子蛋白認得。(3)
Prion在細胞膜上的生成與散佈情形:
(8)
Prion Protein的生物特性:
Prion Particle
Prion被證實全部是由在異常環境下, 構形發生變化的蛋白所組成
. 因此可以這麼說, 所謂prion是一種感染性蛋白. 根據Pan等學者1993年的研究指出, 正常細胞的PrP(以PrPc示之) 轉變為致病的PrPsc乃是經由二次結構的改變,
其中a螺旋和 coil構造轉變為s折板. 這樣結構上的改變也整個影響了PrP在生理化學上的性質, 如PrPc在清潔劑中溶解但PrPsc不會, 而PrPc
易於被蛋白酵素分解, 但PrPsc卻有部份抗性.
對於prion所呈現的不同性質, 我們知道PrP是以內鑄的基因序列存在於染色體內,
代表不同的基因組合能提供prion不同的特性 (strain properties). 但除內鑄基因不同之外, PrPsc三次結構也含有許多具物種專一性(strain-specific)的特性.
僅管PrPsc是目前唯一已知的prion成份, 但在某方面,
prion所呈現的表現型(phenotype)與病毒也十分類似. 因此僅管許多的研究結果出爐, 仍然有部份學者對prion的成份有所質疑.
Molecular Genetics of Prion
Diseases
PrP基因的突變在遺傳上與家族性prion疾病有關. 在Bueler等學者1993年的研究中發現,
缺失PrP基因(Prnp0/0)的老鼠無法複製PrP, 同時對prion的感染具抗性. 由此更進一步地證實, PrP在prion 疾病傳染的過程中扮演重要的角色.
Scrapie在老鼠的潛伏期被用以區別prion品系, 潛伏期的長度是由基因所控制的,
被稱為Sinc基因.
Prion Protein Structure
由於整個PrP是由完整的exon所轉譯, 因此不可能為兩種PrP的isoform經由mRNA
splicing所組成. 至於轉譯後的修飾也可能造成PrPc和PrPsc間的差異, 但目前並無研究證實. 故PrPc和PrPsc間的差別可能只在於其構形上的改變.
兩種PrP的isoform皆攜帶GPI分子, GPI連接的PrPc在細胞表面顯現出免疫反應.
PrPsc模式顯示導致疾病的isoform含有refolding的區段, 位於s折板108-144殘基處. 在這幾個二次結構發生缺失情況下, 將使PrPsc無法形成.
Prion Replication
在未受感染的細胞中, 含有wild-type序列的PrPc可能與X蛋白或與其a螺旋單體,
protein-sensitive狀態同時處於平衡態. 與X蛋白結合的PrPc構形則稱為PrP'. PrP'與X蛋白的複合體將與 PrPsc結合, 形成具複製能力的集合體.
PrP發生構形上的改變, 而變成能和X蛋白結合的形狀, 代表感染性的PrPsc初步形成. 值得注意的是基因重組形成的PrP能自行fold成a螺旋或s折板,
但兩者皆不具有感染性.
各種證具皆顯示PrPsc在動力學上較PrPc穩定, 因此可避免在正常狀態下形成PrPsc.
Prion遺傳學研究
Prion到底是如何形成的? Prusiner等學者認為這是一種由鑄模導引的蛋白折疊,所謂的template-direct
redolding模式. 更進一步地, 這種過程可能由一種類似chaperone分子的protein X協助. 另一方面Gajdusek與Lansbury等學者認為PrPc與PrPsc兩者在溶液中是存在於熱力學平衡的狀態下.
原本PrPsc以單體存在 (正常狀態),經由結晶作用使的PrPsc多聚物產生(病理狀態)--也就是nucleation模式. 目前的各項實驗結果仍然無法確實證
明這兩種假設的真誤. 而也有學者提出, PrPsc可能是PrPc在合成 時的中間folding產物, 然而PrPc的folding過程非常之快, 這樣的解
釋因此尚有爭議.
然而, 在結構方面的研究發現某些PRNP突變種與野生種的具有相同的穩定度.
這顯示PrPc的去穩定性既不是一種一般性的機制, 也不是造成人類TSE表現型的原因.
目前所知, 除了Prnp基因(圖一)之外, 在人類並沒有其它調控prion疾病的
遺傳因子. 然而較可能的情況是許多基因控制著與prion感染性有關 的因子, 如關於淋巴球的成熟等等.
利用transgenic mice進行的實驗, 使得對於Prnp基因與prion疾病的表現方面有更進一步的了解.
Prnp基因缺失(Prnp knock-out)的老鼠已知對prion感染似乎具免疫力. 在prion品系的研究上, 含有不同 Prnp基因並不代表能建立確實的種族屏障(species
barrier). 如 BSE可能在忽略個體間Prnp差異的情況下仍能進行傳染. 另一方面就診斷 方面而言, 經實驗得知在hemizygous
Prnp(只有一個Prnp對偶基因具活性)的老鼠中, 也就是腦中只有半數正常的PrPc含量的情況下, 其發病的 時間要較野生種來的慢.
在vCJD病例中, 最令人值得注意的是影響個體對BSE感染性的因子:
在 vCJD患者中, 並沒有特定的一群與曝露在BSE病原的機率高低有關(如牧場工人, 肉品加工者). 這意謂著有一種潛在的因素影響著個體對於prion的感染性,
可能是由於其基因上的特徵使得對於prion感染有不同的表現型.(4)

★ 人類的prion蛋白質的253個胺基酸順序及其基因突變的位置。套色之處(M129V)是正常人就有的情況(polymorphism)。
在接下來的這個網站,介紹了PrPc和PrPsc的3D立體結構。從鼠類類E.coli中找到,PrPc無β-sheet結構,PrPsc則有。文中亦提到腫瘤突變p53會associated病變protion,並induced正常protein的conformation改變,成為mutation
form,這跟prion的致病力類似。
下兩圖show出以蒼鼠的prion
disease protein圖,構築human prion disease的mutation圖,以及可造成PrPc二級結構disrupt的位點。

(10)

(10)
Prion(傳播性蛋白)的角色:
由於動物及人類傳播性海棉樣腦症可經由疾病腦組織萃取物的注射傳播給其它動物的大腦,因而幾十年來被視為是一種慢病毒(slow virus)感染的結果。但在倫敦Hammersmith醫院,Tikvak Alper的研究首先證明羊搔癢病的大腦經紫外線或離子放射線處理後仍具有傳播性,因而確立其致病物質不含核酸(nucleic
acid)。而不具DNA及RNA之傳播性物質應該不是濾過性病毒或目前已知的其他生物質。
美國UCSF的Stanley B. Prusiner從1974年開始純化羊搔癢病腦的傳播物質,而於1982年純化成功,1984年正式發表Prions。Prions原意為蛋白感染性粒子(proteinaceous
infectious particles),被證明是可以遺傳或傳播這些傳播性海棉樣腦症的蛋白物質。被純化與證明存在的Prions不含核酸,主要成份為蛋白質,因而尋找其細胞體上的控制基因成為很重要的工作。
羊搔癢病的Prion(簡稱SPr)含一個單一蛋白稱為SPrP,在1984年證明其一端有15個胺基酸並確知其排序,而後設計出分子探針,偵測動物細胞是否攜帶PrP基因,並成功的找到這個基因。
在當時發覺所有動物染色體上的PrP基因,可製造PrP但不會致病,因而假設PrP有兩種。後來證明正常的PrP可由細胞內的酵素代謝,稱為酵素敏感蛋白(Protease-sensitive
PrP),而另一種致病的PrP,包括SPrP,皆為酵素阻抗蛋白(Protease-resistaut PrP)。
1986年起經由遺傳性人類傳播性海棉樣腦症庫賈氏病(Creutzfeldt-Jakob
disease,CJD)提供PrP基因研究之機會,在1988年這從Gerstmann-Strarssler-Schneinker病病人身上成功地選殖(clones)出PrP的基因。
基因是由兩鏈(strands)的DNA所組成,而DNA則由帶有不同基(bases)的核甘酸(nucleotides)構成。在DNA其中一鏈的基會和另一鏈上的基做配對結合,而形成所謂的基對(base
pairs),亦即眾所熟知的DNA梯(DNA ladder)。基對除了構成DNA梯上的橫木以連結兩鏈之外,亦負責決定胺基酸序列以製造某一特殊蛋白質。三個連續的基成為一單位稱為密碼子(codon),負責轉譯出一特定的胺基酸。而1988年這名病人身上只有一個基發生改變,造成密碼子102所攜帶的訊息改變,而使其PrP蛋白內的胺基酸leucine被proline所取代。接著,在其他病人及家屬亦發現有同樣的基因突變。至此終於建立基因突變與Prion疾病之關連。在過去六年來,在遺傳性Prion病的家屬中已發現18種突變,其中5種已證明其基因關連性。目前也已成功地改變小鼠的基因,使它們攜帶突變的SPrP基因,且可傳播Prion疾病。至於正常的PrP與SPrP有何不同,正常的蛋白由αhelices所組成,其中蛋白的骨架轉成特殊的螺旋狀,而SPrP包含β鏈(strand),其使其骨架完全撐開或呈折枝狀。這些β鏈形成β薄板。至於增殖的SPrP如何損害細胞,仍有待進一步研究。但目前已知道SPrP累積在神經細胞的溶小體(lysosome),會自己吞噬細胞致死,而留下腦組織之空洞。這些由SPrP會造成繼續攻擊其他神經細胞。當然SPrP亦可在細胞外形成如阿茲海默氏症病一樣的斑塊(plaques),它是診斷Prion病很好的根據。
正常PrP轉變為SPrP,在同種不同部位、或不同種動物可能產生不同之影響,因而從1980年代起大家重新思考種族屏障(species
barrier)的問題。因為種族屏障可以解釋為何一種動物的異常PrP較難引起另一種系動物之Prion疾病,特別在英國發表流行性狂牛症後,此問題顯得極有興趣。
種族屏障是60年代Pattison研究羊搔癢病很難傳播給囓齒動物而發現的。最近在Prusiner的實驗研究也證明小鼠與hamster在254之16
codons之PrP也不同,正常小鼠接種hamster之異常PrP很難得病,但transgenic小鼠則會得Prion病。進一步實驗研究證實所謂種族屏障主要為PrP之胺基酸排列,SPrP分子排列越像宿主之PrP排列者,越易得到Prion病,反之則較難。因為牛的PrP與人的分子差很多,至少30個位置有異,因而牛傳播給人應該是較難的。當然有些異常PrP分子的位置,可能較易打破種族屏障則為人畜共通傳播提供另一可能之解釋。(5)
Prion的傳染途徑:
先天遺傳並不常見,是體染色體的遺傳(autosomal),且為顯性(dominant)遺傳。Kuru
是以前存在在新幾內亞某些土著部落的疾病,疾病經由食用去世親友的腦部而得到傳染。最初在 1950年代被發現,最後經由禁止這種風俗而停止這種疾病,但是其他部落有相同習俗的,也沒有事,應該是這個部落特有的遺傳疾病,後來經由食用去世親友的腦部而傳染。
後天感染則是接受了已被感染的物質,例如食物,是食用了含有病原體的肉類、或是注射、或器官移植等。羊也可以因為吃下了初生小羊的胞衣而得病,在英國的瘋牛症,是因為使用了銷毀的羊肉製品如骨粉等,餵食牛以補充營養而得到感染。有不少報導警告,人若食用了罹患瘋牛症的牛肉,也有機會得到 CJD,因為這是大多數患者感染的途徑。
普恩蛋白質雖然只是一種小小的蛋白質,但是卻是到目前為止,最頑劣的生物分子,我們至今仍然不明白將其消滅的方法,未來還不知道要經過多少人的努力,才能使我們脫離受普恩蛋白質襲擊的恐懼?
自從愛滋病的病原體被發現之後,人類對所有新發現的病原體都非常的恐懼,其實,普恩蛋白質並不是病毒,或許稱之為病原素、或病原質,會比叫它病原體更合適。但無論如何,這一種新病原已經引起了人類的「新病原恐懼症」。
在英國,庫雅氏症致命的個案已經有迅速上升的趨勢。英國去年有十四人死於庫雅氏症,但在今年首六個半月已有十四人喪生,另有五人奄奄一息。牛津大學動物學家安德森估計,如果這個趨勢持續,則未來三十年將有多達五十萬英國人,死於這種透過進食受感染牛肉製品所傳播的疾病。
曾獲諾貝爾獎的科學家布魯希納警告,瘋牛症已經成為英國羊隻流行的夾病了,普恩蛋白質已經在英國羊之群間廣泛散播。他說:「我們新近進行的研究顯示,瘋牛症已成為英國全國羊隻的流行病。」
布魯希納的研究指出,所有哺乳類動物的腦部都含有這種蛋白質,但是它可以自己改變結構,形成有害的普恩蛋白質,繼而產生連鎖反應,引起瘋牛症等數種可致命的腦部疾病。如果吃了受感染的動物,將出現同樣的連鎖反應。
過去科學家從未在綿羊身上發現可以導致瘋牛症的有害蛋白。不過,布魯希納和研究人員最近做實驗,發現老鼠接觸到患有羊搔癢症羊隻的物質時,老鼠竟然出現瘋牛症病徵,顯示羊隻可能帶有引起瘋牛症的普恩蛋白質。
瘋牛症也是一種傳染性海綿樣腦病變,於1986年在英國發現後,至少造成16萬5千頭牛的死亡。在八十年代中期,英國曾有大批的牛隻因為感染神秘的腦病而相繼病倒。研究人員追查之後發現,問題在於牛隻的蛋白質飼料,是由受感染動物的腦部及神經組織所提煉的,牛隻可能是吃了感染羊搔癢症的羊隻所作成的肉骨糜飼料之故。
瘋牛症的潛伏期會超過二十五年,到目前為止,曾經接觸過受感染之牛肉的人,數以百萬計,但有多少人確實被傳染,則尚未證實。也有科學家表示,英國的瘋牛症可能已透過被污染的嬰兒食品、和學校提供的營養午餐傳播開的。
有人說:「以前所謂的不治之症,只是一些我們不懂得底細的病」,懂了病的底細,將來才會有治療的對策,布魯希納証明了這句話。他是一位遠赴人跡未到的自然界的一個死角,把沒人料到的新型病原體從其藏匿處揪出來示眾的人。(2)
由於同一種prion的感染會造成不一樣的症狀,因此當初有人認為這種感染是病毒,因為病毒含有核酸而會有多株的產生。現在已知道這是prion有多種的形態結構所造成的,不同型態的普恩蛋白其複製的時間及影響神經元的部位都不太相同,也因此產生臨床上不一樣的症狀。(19)
※ 在未來對prion的研究將一日千里,一定能找到完善的調控prion的方法,以避免人類浩劫,以及經濟產物的恐慌。
Prion發現早期認為可能由病牛傳播到人體的路徑:

(9)
以毒攻毒
物理學家透過電腦模擬提出一種對抗狂牛症的新方法-以毒攻毒。
狂牛症是經由一種普恩蛋白(prion)所引起的神經異常疾病。由於對動物的影響及另一種Creutzfeldt-Jacob疾病(簡稱CJD)對人類的影響,這種由普恩蛋白所引起的疾病已經成為人類的嚴重威脅。
美國UC Davis的兩位物理學家Cox和Singh用簡單的統計力學模型來模擬了普恩蛋白疾病的傳播。他們利用一組二維的晶格系統來進行模擬。他們發現一點點的prion可以在受到感染的神經元上製造出更多的prion。當到達足夠的數量之後便會分裂而讓其他的prion傳播出去到別的神經元上,最後造成宿主的死亡。
事實上prion具有幾種不同的種類,例如有些對老鼠及倉鼠造成傷害但有些種類則是對倉鼠無害。這個模型發現,若是將無害的倉鼠普恩蛋白注入受感染的老鼠身上,可以使兩種不同的普恩蛋白彼此競爭,如此一來便可能可以減緩發病的時間。這種以毒攻毒的方法並不能保證能消滅這種疾病,但卻可以使類似CJD這種疾病不易在人身上發作。(7)
因為引起CJD疾病的蛋白質是一傳染性極高、非常穩定、不易被破壞的分子。因此,對於實驗室安全操作、管理、緊急措施、廢物處理等等一定得訂定遵循之規範。由實驗室診斷上,確認是否是新變種及散發型之CJD疾病,仍需進行糖化分子之分型與腦之組織切片。但因這兩者使用之材料,必須直接取自腦組織,故具有極高之傳染性。因此也需要在極高安全等級之實驗室進行。國內將來將建立相關之實驗室,將可執行進一步之新變種及散發型之CJD疾病之鑑別診斷。(21)
目前雖無跡象證明人類CJD疾病會經輸血途徑傳播,但其真正治病機制尚未十分明瞭,以知尚涉及先天因素在內,正常普恩蛋白第129胺基酸序列若為methionine同型結合子,則為CJD疾病易感者,易感者可能一旦有低濃度病媒接觸,亦於致病。加上近年英倫發現新變異型的病例,認為與前不久英國爆發的狂牛病有關,傳統CJD疾病經血液傳染的不確定性,同時由CJD疾病異常普恩蛋白的致病機制研究所見,使人擔憂近年日漸興起的基因改造食物,台灣日常食用的黃豆、玉米及各種豆類食品皆是基因改造農產品,此種加入動物基因改造的植物,違反自然,破壞生態。長期食用及一旦遭遇先天遺傳易感者,有可能會造成類似CJD疾病相同致病機制的病症,則將成為人類一場大浩劫,醫界莫不戰戰兢兢,嚴陣以待呢!(17)
我找到有一本書專指介紹prion蛋白的研究,內容揭露出一種不知名的致命疾病,其致病因子就潛藏在人類所食用的肉類當中,它會破壞人體的免疫系統,蝕空頭腦組織,癱瘓說、聽、走、吞食能力,最後使人只求一死,除了成千上萬的牛和家畜死於這種疾病,連英國及法國也有年輕人遭到感染,未來美國大陸及其他地區也極可能無法倖免逃過被傳染。已知的狂牛病就是這種致命疾病之一。本書作者跟隨科學家(包括本屆諾貝爾生醫獎得主布魯希那),深入非洲熱帶雨林,追蹤這種神秘疾病的歷史來源及傳染情形,希望找出預防爆發傳染之安全措施。如果不加以遏阻,此病有可能比世紀黑死病更可怕。本書之出版將引發對食物安全爭論及科技危機,震撼人心,卻能挽救人命。
書名:致命的盛宴
作者:理查‧羅德斯
譯者:汪仲,張定綺
出版社:時報出版
初版日期:1997 年 12 月 23 日
(11)
補充資料:
Prion之相關研究:
1976年諾貝爾醫學獎得主
Daniel Carleton Gajdusek
Daniel Carleton Gajdusek, 1923年9月9日出生於Yonkers,New
York。父母親是來自東歐的移民,以經營肉舖為生。自小就對科學有濃厚的興趣,也對詩文、音樂及其他藝術頗為傾心。少年時期在昆蟲學家Dobrosky姨媽的引導,及一位數學及物理化學家Dr.
Youden的啟蒙下進入科學研究的殿堂。十歲時,對於自己準備將來要達成學習醫學的志願時為什麼捨古典生物學而先學習數學、化學、物理的原因寫成一篇隨筆,提到了那是因為Dr.
Youden教導他數學、物理、化學為生物的基礎。
1940到1943年,在Rochester大學學習物理、生物、化學及數學。課餘時亦熱衷於登山、郊遊、划獨木舟、露營。隨後便到Harvard
Medical School就讀,同時參與蛋白質物理化學,病毒學的研究。其間曾入迷於小兒科臨床醫學並取得小兒專科醫師資格。1951年奉召服兵役,從Harvard的John
Enders(亦為諾貝爾獎得主)研究室,以病毒學家被調到Walter Reed Army Medical Service Graduate
School服務。在Dr. Smadel的教導下,學習實驗室與野外研究的方法及研究結果發表的方法。從此到達世界許多角落,從事病毒學及流行性疫病的研究。
‧ 由Vincent Zigas引薦Kuru的研究‧
1954年起服務於澳州墨爾本的研究機構,當時他曾聽說過Kuru。一直到1957年,在一個偶幾天的相處下,極受贊賞而被邀請參與此項研究工作。從此便廣泛且深入的研究Kuru,後來由於證實了Kuru係由slow
virus infection致病而於1976年獲得諾貝爾醫學獎。
‧ 什麼是Kuru?‧
Kuru為Fore族語,是顫抖的意思。此病發生於新幾內亞東方高地的Fore族。染病者為婦人與小孩,漸進性的消瘦,步態不穩,不自主的顫抖、痙笑,一年內致死。當時在澳洲以Bennett為首的研究小組,認定這是一種遺傳家族性疾病,甚而興起欲築起一道高牆將疫區與外界阻隔,禁止通婚的計劃。但是Gajdusek等在走訪超過40個病例,做了非常詳細的流行病學與臨床觀察後,在1957年發表他們的見解於新英格蘭醫學雜誌。接著再經過仔細分析研究後,雖然也無法排除遺傳家族性致病的可能,但卻在另一方面一直積極又廣泛的尋找檢驗致病的因素。同時也注意到英國獸醫病理學家Hadlow於1959年在Lancet的論文“Scrapie與Kuru”中提到Kuru的臨床及病理發現與Scrapie非常的相似,從而又回想到當時流行病學所發現Fore族的一大特點,即有食人肉的習性,而導引其研究方向於以實驗方式,將Kuru死者之腦取液注射於各種小型實驗動物之腦內,開始時卻都失敗。
‧ Scrapie又是什麼?‧
Scrapie流行於英國及歐洲大陸已有200多年歷史,為發生於羊的一種漸進性神經系統退化症。其症狀為步態失衡,顫抖,終而死亡。在病程中會以身體到處刮擦,因而稱為Scrapie。其病程中並無感染症狀,而且在腦部的病理變化亦無炎性反應,最突出的變化是大腦皮質呈海棉般鬆散空泡(海棉樣腦病)。早在1899年已有學者進行傳染實驗。此研究一直到1936年又再度被證實可傳染。
1956年,Gajdusek將得自11個Kuru病人的腦瘁取液注於十幾隻大猩猩的腦內,18-38個月後,大猩猩們開始發生與Kuru臨床相似的病症,至此首次證實Kuru為傳染性的疾病。1968年此結果發表於Science上。以後幾年,他的研究群又致力於將發生於人類的一些慢性神經病變的腦瘁取液注射於大猩猩及其他實驗動物身上。結果只發現Creutzfeldt-Jakob
disease(CJD)也同樣能傳染。
‧ 從Kuru經CJD到Alzheimer病?‧
CJD係發生於中老年人,主要症狀為早發性癡呆併不自主抖動,平衡障礙等,往往在發病後一年內死亡。其病理變化為與Scrapie,Kuru相似的海棉樣腦病。雖然他們證實了上述這些病具有慢性傳染性,接續的問題便是此傳染物質到底是什麼東西?早期的看法,認為這類疾病是一種遲發性感染Slow
infection,為了與一般病毒感染區分,最常被稱呼的是Slow virus infection。由於其傳染物質一直未能被分離辨識因此又被歸屬為非傳統病毒。
進一步重大的突破為1982年由美國加州大學的一位年輕神經學教授Stanley
Prusiner發現Scrapie之傳染物質為一種蛋白質Scrapie-specificamyloid protein而稱之“Prion”protein。目前發現Scrapie, Kuru, CJD病理變化上的澱粉樣斑(amyloid
plaques)均與Prion protein有關。Gajdusek的研究小組除了持續的以分子遺傳學方式探討散發性及家族性CJD的致病機序外,由於Alzheimer老人癡呆症的病理變化亦有澱粉樣斑,而引發他們做進一步比較研究,以期揭發老人癡呆症的致病機轉。(6)
★ 在動物的防疫上被歸類為乙類動物傳染病
http://www.baphiq.gov.tw/animal/disease/disease.htm
★ 詳細狂牛症、綿羊搔癢病以及CJD報告,上此網站可查詢
http://www.mad-cow.org/~tom/
http://nr.stic.gov.tw/ejournal/scipolicy/Sr9004/SR9004T3.HTM
http://members.tripodasia.com.tw/Annika_Tao/articles/performance-1c.htm
http://wwwo.hkedcity.net/public/ileisure/research/200106/15673.phtml
http://www.vghtpe.gov.tw/~neur/neurology/gn/chapt1.htm
http://www.vghtpe.gov.tw/doc_vgh/gn000a.htm
http://www.coa.gov.tw/magazine/farming/9003/015.htm
參考資料:
1.
http://microbiology.scu.edu.tw/micro/people/Prusiner.htm
2.
http://www.twbm.com/chinese5/science_c4.htm
3.
http://vm.nthu.edu.tw/science/hall/
4.
http://www.geocities.com/Athens/Styx/6382/arttext2.htm
5.
http://bbs.nsysu.edu.tw/txtVersion/treasure/neurology/M.889933960.A/M.890305325.A/M.891230765.F.html
6.
http://www.medlib.ncku.edu.tw/people/1976.html
7.
http://www.bio.idv.tw/paper/16.htm
8.
http://www.rkm.com.au/BSE/
9.
http://www.rkm.com.au/BSE/
10.
http://www-micro.msb.le.ac.uk/335/Prions.html
11.
作者:理查‧羅德斯 譯者:汪仲,張定綺 致命的盛宴 1997 年 12 月 23 日
時報出版
12.
http://www.mad-cow.org/~tom/braingifs.html
13.
http://www.nobelchannel.com/prizes/przw_vid.sps?id=160&category=Medicine&year=1997
14.
林培正 具有傳染性的蛋白質—普恩蛋白 中華民國83年9月 科學月刊第二十五卷第九期 P.679~685
15.
白美蜀 傳奇的普恩蛋白之新發現 中華民國84年8月 科學月刊第二十六卷第八期
P.639~646
16.
陳秀雯 Prion及狂牛病 1998年3月15日 食品工業月刊第30卷第3期 P.11~20
17.
王玉祥 輸血是否會傳染狂牛病 2001 台灣醫界Vol.44,No.10 P.25~27
18.
劉錦志 獸醫病毒(一)現代分子病毒–PRION 民國86年12月 氰月安牧苑第十六卷第三期 P.4~7
19.
翁逸豪
林佼瑩 楊振典 普利子疾病(Prion Disease) 民國87年01月 台灣醫界41卷第1期 P.28~30
20.
呂建榮 Prion疾病 中華民國八十八年四月 當代醫界第二十六卷第四期 P.312~315
21.
李進成
劉振軒 王傳亨 傳播性海綿狀腦病變的病理變化與分子診斷 2001年 台灣醫學5卷5期 P.562~568
22.
http://cmp.ucsf.edu/cohen/research/gallery/aw_prion.gif
|