蛋白質結構模擬原理
口述:黃明經 老師
整理:李京倫、黃淳瑩、卜峰麟
b.摺疊辨識法(Protein fold recognition)
1. 一維-三維側寫法(The 1d-3d profile-method)
1.運用I-SITES, HMMSTR 及ROSETTA 來預測蛋白質結構
2.1微秒分子動力學模擬(1-microsecond MD simulation)
隨著後基因體時代的來臨,人類對於基因的研究焦點,已由解碼定序轉移到基因表現的最終產物----「蛋白質」上﹔儘管科學家得知基因的序列,但還必須進一步了解此基因所隱含的生理功能﹔由中心法則(central dogma, DNA->RNA->Protein),基因最終以蛋白質的形式表現,蛋白質的功能則與其三度空間結構有密切關係﹔因此,由蛋白質結構探討其功能是未來的重要方向,在蛋白質結構尚無法藉由現有技術全部解構出來之前,結構的預測與模擬則是探討生物分子功能的重要研究領域。
在蛋白質結構上,有所謂的一級決定三級,即胺基酸序列可以決定一個蛋白質的結構﹔但由蛋白質結構資料庫(Protein Data Bank, PDB)所儲存的結構來看,大多數蛋白質結構的影響因素並非只有胺基酸序列而已,因此仍無法直接由胺基酸序列來模擬三級結構﹔蛋白質結構主要仍是由實驗的方法來取得,但以實驗的方式來找出每一個蛋白質結構,則需要時間,主要是因蛋白質的純化及結晶具有一定的困難度。蛋白質可以經過定序,幾天內就能得到完整序列,但是要解出其結構則可能需要數年的時間,因此在序列和結構之間就出現了鴻溝,仍有待克服。
蛋白質結構在不同環境(溫度、pH值、離子濃度)下,其構形會產生些微的改變﹔不過每一種蛋白質皆有某一具有功能的構形,若蛋白質沒有摺疊成它正確的形狀,會使得蛋白質的功能無法發揮,有時這種摺疊錯誤的蛋白質還會對生物體造成傷害﹔例如狂牛症的發生,即是原具正常功能的蛋白質-Prion,經點突變造成結構上的改變所造成。而更有趣的是,某些蛋白質它天生就是不需摺疊,一旦摺疊起來反而會喪失其功能。
在理論上,可以利用模擬的方式來預測巨分子的結構,有名的例子如:Watson和Crick決定DNA 結構﹔他們並沒有實際做過實驗,但是憑藉其他實驗室所得的數據和X光繞射圖譜,大膽預測了遺傳物質DNA為雙股螺旋結構模型,並以此獲得1962年諾貝爾生理醫學獎;而Linus Pauling則利用分子模型來模擬α-helix,而獲得1954年化學獎。
目前對於蛋白質結構的研究方法,主要可分為兩大類:一是利用實驗的方法,包括以X光繞射(X-ray diffraction)或用核磁共振(NMR)物理的方式來建立蛋白質的結構模型;另一種則是利用電腦的計算,依據現有的結構資訊配合理論運算,進行未知結構的預測﹔預測的方法則包括了同源模擬法(Homology modeling)、摺疊辨識法(Folding recognition)以及重頭起算法(Ab initio)三種。以下將針對這些結構研究法做介紹:1
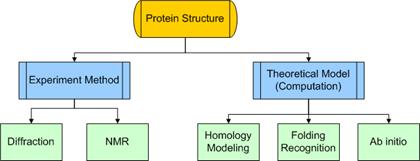
【圖1.蛋白質結構研究方式歸類】
a. X光繞射(X-ray diffraction):利用光線的繞射特性,對形成晶體的蛋白質以X光束照射後產生繞射圖譜,再將此平面圖譜,經由一連串的計算分析,並取得晶體的像角(phase angle)後,再加以建立蛋白質結構模型﹔本法是目前蛋白質三維結構的主要來源,但其缺點是,約有百分之五十的蛋白質不易取得晶體,其純化的過程亦有其難度,需要更進一步的改進表達系統(expression system)及結晶方法,以加速蛋白質結構的建立。
b. 核磁共振(Nuclear Magnetic Resonance, NMR):利用電子的自旋(Spin)的特性,以外加磁場改變其自旋方式,藉此獲得蛋白質的電子圖譜,依取得之數值計算蛋白質結構。因NMR所解出來的結構,係蛋白質在水溶液中之狀態,屬於動態模擬方式,可以得到20~40種模型,再經由運算,取得平均結構﹔由NMR的結果,可以模擬蛋白質在生體內(in vivo)的動態結構﹔但因共振儀及訊號強度的限制,NMR所能解出的蛋白質結構約在分子量10 kd,約為100個胺基酸,所以蛋白質的長度是其限制,若能提昇其解析能力,對蛋白質結構的取得亦可快速增加。
a. 同源模擬法(Homology modeling):將目標蛋白質序列與現存於蛋白質結構資料庫(PDB)已知序列進行比對,尋找出最好的三維結構模板(Template),以此模板為模型將目標序列導入後,取得三維結構模型,經分子動能(molecular dynamics)及能量最小化(energy minimization)運算,以獲得目標序列的蛋白質結構。此方法的缺點是當現存資料庫中沒有類似之結構序列或序列相似度較低(<30%)時,就無法以此一方法進行預測,即使強行利用此法模擬,其預測的結果可信度也不高。
b. 摺疊辨識法(Fold recognition):將目標序列分區段,代入已知蛋白質結構序列的相似摺疊模板,觀察二者在某一區域內之相似度,依序列順序跟結構排列的法則,經能量最小化,來找出符合要求的分子。本法需耗費大量的計算資源,對於蛋白質的核心部份具有較高的準確度,其外圍的二級結構則較差。
c. 重頭起算法(ab initio):以分子動力學的原理,考慮胺基酸和溶液的所有交互作用力,找出分子間最穩定的狀態﹔由一級結構開始,來計算出蛋白質的三級結構。此一預測法同樣需要大量計算,且只能對短鏈胜肽(peptide level)進行預測。
綜合以上所介紹的蛋白質結構產生方式,以實驗方法來進行的模型建立,其共同的缺點便是太過耗時,且常受限於樣本的製備技術。而欲分析的蛋白質若無法透過實驗方式取得三維結構時,要如何了解蛋白質受質結合區域或蛋白質-蛋白質交互作用區域?因此,在蛋白質尚未以實驗方式取得結構模型前,就必須以結構模擬的方式來進行﹔目前較理想的方法是分析蛋白質序列,由現存結構資料庫中取得原子三維座標,透過電腦運算建造蛋白質的結構模型,經結構最佳化,並評估模擬蛋白質結構,以取得其三維結構模型。2
接下來即是針對理論模型所簡介的三種產生模擬蛋白質結構的方式,做進一步的介紹:
又稱為比較性模擬法(comparative modeling)或知識基礎模擬法(knowledge-base modeling),利用本法可以快速模擬蛋白質的結構。它主要的方式是利用現存已解出的結構為模板,模擬出目標蛋白質分子結構。一般而言,蛋白質序列和模板序列之間的相似性越高,所模擬出來的蛋白質結構其正確性也就越高。
步驟1:選擇和目標蛋白質分子相關的其他參考蛋白質分子
依現有蛋白質結構與序列間的關係,若二種蛋白質間的序列相似度大於30%以上,則表示二者之間的結構具有相當高的相似性,同時也表示二者之間的功能非常接近,因此使用同源模擬的方法預測未知蛋白質結構,是具有其一定的理論基礎。所以,只要同源蛋白質家族中其中某一蛋白質,經由實驗方法決定其結構後,就可以利用同源模擬的方法來預測其他一些序列已知而結構未知的同源蛋白質分子結構。但若是目標蛋白質與現有具有結構之蛋白質序列,二者相似度低於30%時,則不適用本法來進行結構模擬,而必須採用二級結構預測、摺疊辨識法或重頭起算法來預測目標蛋白質的結構。
步驟2:比對目標蛋白質和參考蛋白質的胺基酸序列
這是整個程序中最關鍵性的步驟,對齊兩條蛋白質的胺基酸序列,即決定目標分子序列中對應於參考蛋白質核心骨幹及環狀(loop)分子鏈的各個段落,對產生正確蛋白質結構有關鍵性的影響。當序列相似程度大於50% 時,兩序列可輕易對齊﹔但相似程度低於50%時,則需一些額外的資料才能得到可靠的對齊序列,困難之處在於確認序列中適當的基準點。
步驟3:畫出目標蛋白質的核心部份
以參考分子的核心部份骨幹結構為基礎,將胺基酸換成目標分子對應位置的胺基酸。核心部份分子骨幹的胺基酸的更換只會改變二級結構元素的相對位置和取向,而不致於破壞這些結構元素或三級摺疊的一般特性。
步驟4:修正最後的分子結構
在產生鬆散部份的分子鏈結構階段可能得到數個合理的分子構形,若不止一段的分子鏈各具有幾種可能的分子構形,則要考慮各段落分子構形的組合構形,再進行整體的結構調整和修正。
步驟5:蛋白質分子結構的驗證
所建立分子模型的正確性必須和該蛋白質已知的實驗數據做一致性的比較來評估,例如:經同源模擬預測所得到的蛋白質結構模型,通常含有一些不合理的原子間接觸,需要對模型進行能量最小化和分子動力學的處理,消除模型中不合理的接觸﹔模型中有些鍵長、鍵角和二面角(dihedral angle),若不符合現有理論值時,也需要檢查評估﹔其他如胺基酸堆積密度、厭水性或帶電荷或極性胺基酸的位置、溶解自由能等實驗測量值,則與已發表的生化實驗證據做進一步的確認。
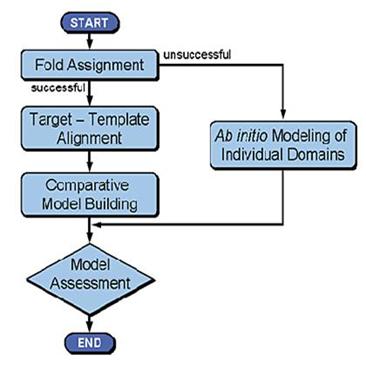
【圖2.由同源模擬法模擬蛋白質解構的步驟】3
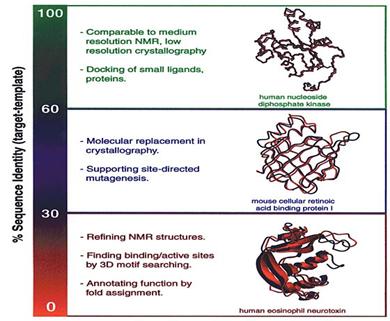
【圖3.目標蛋白質和參考性蛋白質序列間的相似度。】4
應用:
同源模擬法主要應用在藥物設計(drug design)、蛋白質特定突變(site-specific mutation)研究或蛋白質活性區域(active site)分析﹔由於以比較同源性的方式,具有相當的可信度,其結構的變異相對較低,因此是目前蛋白質結構模擬中,最常使用的方法。
同源模擬法的限制:
1. 在預測蛋白質時,同類蛋白質至少需有一種晶體結構或核磁共振結構已知。
2. 蛋白質結構資料庫中可供參考的蛋白質結構仍有限。
3. 在同源性很低時,兩胺基酸序列比對,以及在環狀次單元(loop subunit)的結構模擬都有待進一步改進和發展。
4. 無法預測蛋白質是否產生新的摺疊。
5. 在簡化位能函數及提供計算效率及處理溶劑效應方面仍有待努力。
6. 經同源模擬法出來的模型可能會偏向模板,而非真正結構。
7. 在預測擁有極高序列相似性的蛋白質其核心構造(core structure)時,結構正確率高,但在預測蛋白質表面構造時,因牽涉外圍胺基酸支鏈與水溶液間的交互作用,其預測結果較易出現不一致的現象。
8. 遇到序列相似度低時,就無法預測或是預測的結果可信度很低。
網站介紹:
http://www.chemcomp.com/feature/bio1999.htm#Search
這個網站介紹了如何利用軟體來做同源模擬法,只要在MOE-SearchPDB中貼上目標蛋白質序列,經過一連串步驟再利用MOE-Homology得到蛋白質3D結構,最後可以得到模擬的目標蛋白質模型。步驟如圖4。
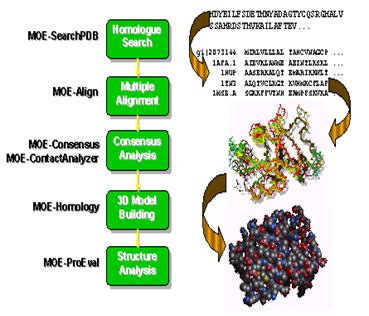
【圖4.在MOE-SearchPDB中作同源模擬的步驟】5
b.摺疊辨識法(Protein fold recognition)
相對於同源模擬需要較高的同源關係外,對於許多蛋白質的胺基酸序列彼此相似度低於30%,但是卻擁有極為類似的三維結構時,則需要採用胺基酸序列在區段同源性較高的部分,來定義區段的摺疊方式,最後再組合成為一個完整的蛋白質結構﹔這就是摺疊辨識法的基本構想,以下便對此方法作詳細介紹:
摺疊辨識法之原理為:將一條序列分段與許多不同的蛋白質結構(或結構的片段)進行比對,計算出此一序列最有可能摺疊成為哪一個結構,將胺基酸序列摺疊成立體結構在空間中位置,藉由空間組合的計分方法,計算不同的排列組合得分,依得分的高低,判斷序列摺疊成為某一立體結構的機率,這種計算序列與結構之間的排序過程稱為穿針引線(Threading)。6根據此一原理,在實際應用層面上,又可分為以下幾個重點:
1. 一維-三維側寫法(The 1d-3d profile-method)
此法為Bowie et al.於1991年首先提出,當時他利用待測蛋白質在18種「環境」(environment)作用的結果來分別描述此一蛋白質不同部位的結構,其後又有許多科學家據此開發出相類似的研究方法(Ouzounis et al., 1993; Yi & Lander, 1994)。在此所提到的「環境」一詞,係指不同蛋白質殘基(residue)間的接觸形式,或是所暴露出的表面積等性質而言。此類方法的共通原則為:
a. 依殘基所在環境狀態,將其三維結構還原為一維的長鏈狀結構。Bowie確認這些環境的方式是藉由量測蛋白質中被包埋起來的支鏈(side chain)面積、暴露於極性原子中的支鏈面積以及局部二級結構。
b. 計分基礎是建立在20種胺基酸在不同環境中的觀察結果,將所得資料與已知蛋白質結構或序列的資料庫比對,相關性越高者分數越高。
c. 依特定位置(position-dependent)在三維結構中的出現機率,建立三維側寫表(3D profile),例如在蛋白質特定位置出現某一胺基酸的可能性。
d. 取一胺基酸序列對上述三維側寫表進行排序(alignment),比對的計分標準則依據此一序列和三維側寫表中所描述的結構相容性高低來判斷。
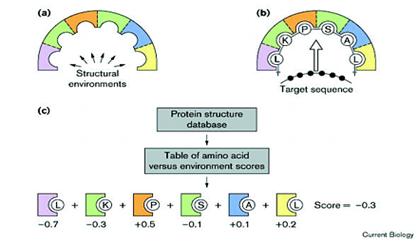
e. 圖5(a)即表示環境中的結構條件,其後如(b)所示,將待測蛋白質的一小部分胜肽鏈與之作用,最後如(c)所示,將觀測數值與資料庫作比對,分別對每一位置做計分評估,找出最高分的組合序列。
【圖5.摺疊辨識法預測原理】7
除了由殘基環境所得到的資訊,有些研究人員尚利用不同胺基酸之間的距離作為預測結構的參數。由於殘基與探針序列特定位置連接時,其所需要的能量大小端看鄰近位置的胺基酸組成為何﹔因此,胺基酸之間的距離資訊,在分子力學方面的分析便提供了有力的計算依據。
蛋白質核心的二級結構,例如組成β-圓桶(barrel)的β摺板,其所形成的摺疊具有特定的性質;因此若胺基酸序列經由預測所產生的二級結構,和經由胺基酸環境所推測的摺疊核心二級結構並不相符時,則該序列不屬於這個摺疊的可能性相對較大﹔反之,若二者相符時,則屬於同一摺疊的機率則提高。
將所有的二級結構元素(Secondary Structure Element)視為一串二級結構字串,則可以使用一般字串排序的方法,將目標序列與資料庫中的二級結構序列排序,除了容忍一定程度的插入,刪除元素之外,還必須符合下面幾條規則,來篩選出較可能的結構:
a. 去除不緊密的結構:蛋白質在自然狀態下會緊密的摺疊,所以分子結構的半徑與其胺基酸鏈長度相關,藉由限制半徑的大小,可以去掉過度伸展,及部分不合理的結構。
b. 環狀長度與距離的限制:連接兩個二級結構元素的環狀部分,其相對應的排序序列長度,與結構上兩端之間的距離,必須有合理的限制。
c. 不合理的二級結構:在排序時雖然可容忍一些插入、刪除元素的操作,但在二級結構中間插入或刪除,會造成不合理狀態,例如孤立的β摺板。
如上所述,用已知的結構作為模板,將相對應的殘基一一取代為目標序列的胺基酸種類,並計算其分數。計分的方式有很多種,下面列舉接觸能量和親水疏水的評分方法:
a. 接觸能量:在空間中位置相鄰,但在胺基酸鏈上序列並不相鄰,我們稱之為有接觸。不同的胺基酸,由於大小、疏水性、帶電量等等性質,兩兩之間接觸所造成的穩定程度不同,在自然界中接觸的頻率也就不同,因此我們可以藉由統計的方式建立出自然界中兩兩胺基酸接觸的趨勢表,用來評估序列與結構排序的結果是否與自然界的趨勢相同,藉此區分排序的好壞.。一般相鄰的定義為,兩個殘基上的α碳原子相距在8 Å之內。
b. 整體疏水性的評估:具親水性的胺基酸,大多暴露在蛋白質的表面部分,而疏水性的胺基酸則趨向於被包覆在蛋白質的內部,群聚在一起。藉由這樣的性質,我們可以評估序列與結構的排序是否仍然維持這樣的特性,若疏水性的殘基卻都對應到結構中的表面部分,則我們可以推測這是一個不好的排序,或是序列並不屬於這個摺疊。
一些關於Fold Recognition蛋白質結構預測Server的網站連結:
1. 3D-pssm:http://www.sbg.bio.ic.ac.uk/~3dpssm/
2. UCLA/DOE Fold Server:http://fold.doe-mbi.ucla.edu/
3. UCSC HMM Applications:http://www.cse.ucsc.edu/research/compbio/HMM-apps/
4. Center of Applied Molecular Engineering:http://www.came.sbg.ac.at/
5. TOPICS (EMBL附屬網頁):http://www.embl-heidelberg.de/
第三種用來模擬出蛋白質結構的方法,是以物理基礎來研究蛋白質摺疊的方法,重頭起算法就是僅由胺基酸序列,而其它的資訊完全無法使用時,來預測蛋白質的三維結構﹔以目前結構的訊息數量來看,尚無法單純由序列準確預測蛋白質結構,尤其在以實驗的方法定出結構之前,是無法評定模擬出的模型其準確程度有多好,但由序列來預測結構仍是可行的。
我們可以評估模擬預測的準確性,不論重頭起算法或是摺疊辨識法抑或是同源模擬法均是以胺基酸序列的資訊來預測蛋白質的結構,但依自然狀態下,蛋白質的摺疊是依照基因轉譯出的胺基酸序列,依序形成具有功能的三維立體構造﹔Anfinsen(1973)發現,在變性的球形蛋白質在回復到原有的狀態時,會自動的再摺疊,回復為原來的球形狀態,因此推論蛋白質摺疊的訊息是隱藏於一級結構(胺基酸序列)中﹔在1994年成立了一個大型的學術團體來評估預測蛋白質結構方法的優缺點,包含了三個部份:
1. 從實驗結果中,預測蛋白質結構。
2. 由晶體結構和NMR所得的蛋白質結構
3. 由發表的晶體結構和NMR模型來預測未知蛋白質結構。
重頭起算法利用對照的方式,藉著比對已知蛋白質結構序列和摺疊狀況來推測出可能的蛋白質摺疊狀況,進而預測蛋白質三維結構﹔重頭起算法以物理學原理來預測蛋白質結構,主要是依據蛋白質摺疊的熱力學定義,此處是假設蛋白質摺疊後,將會使其能量逹到最低的狀態。以短鏈胜肽所摺疊出的二級結構,來推測完整蛋白質經摺疊後所產生的結構﹔此模擬包含了利用一個或數個殘基交互作用中心(residue interaction center)及以晶格描述蛋白質法(lattice representation protein)。
預測局部性結構分配的位置可以使用HMMSTR(hidden Markov model for local sequence-sequence motif)﹔此方法亦可以應用於預測無重覆(non-redundant)序列的蛋白質結構的。
1.運用I-SITES, HMMSTR 及ROSETTA 來預測蛋白質結構
預測蛋白質結構,可使用接觸圖譜(contact map)的方法,其主要內容係以局部結構狀態的多樣性以及結構的規律性來獲得﹔經由預測可能的局部性的蛋白質結構,再藉此來預測可能的蛋白質接觸圖譜,由此經驗法則來預測蛋白質摺疊後的可能結構。使用HMMSTR可以得知,每一種區塊(motif)將有一特定的結構,為滿足前後相關的HMMSTR狀態下,及無重覆結構的需求,將使用三種接觸圖譜﹔1.穿針引線法:調整以HMMSTR為目標的模板,每一個模板以具接觸潛力的計算值作比較。2. 序列同源性:如果穿針引線不是決定性的步驟,我們需尋找具有高相似性的模板,使其和許多模板比較,此種方法可以預測新的摺疊方法。3.重頭起算法:當穿針引線法及序列同源性不足以使用時,就使用物理學的方法來建立接觸圖譜。
2.1微秒分子動力學模擬(1-microsecond MD simulation):利用分子動力學(MD, molecular dynamics)的方式,找到一個最穩定的狀態,可能就是蛋白質摺疊後的狀態,而分子動力學要分成許多方面來探討:1.模型系統、2.起始狀態、3.分界狀態、4.整合計算、5.限制條件、6.組合、7.結果
分子動力學循環及整合計算的策略可以分為下列幾個步驟: 1.計算蛋白質能量、2.計算每個原子的作用力、3.計算每個原子加速度(acceleration)、4.計算每個原子的速度、5.計算每個原子空間位置。
3.時間度量
在不同的時間,整個蛋白質會有不同的摺疊狀態,依此來計算最低的能量狀態﹔為找出那一個是最小能量狀態的摺疊,將以下列幾種不同的時間度量情形來探討:
鍵結原子的相對振動à10-14秒
蛋白質表面的胺基酸支鏈旋轉à10-11秒、10-10秒
包埋區域的扭轉振動à10-11秒、10-9秒
不同球形區域的相對移動à10-11秒、10-7秒
蛋白質內部之中型胺基酸支鏈之旋轉à10-4秒、-1秒
蛋白質本身的變性à10-5秒、-10秒
分子機制與動力學的組合需要考慮到下列三種因素:
由統計力學中的microcanonical(N.V.E)及canonical(N.V.T),可以使得原子路徑的軌跡,以積分的形式作運算,導入重頭起算法中﹔固定壓力(constant pressure, N.P.T)在一大氣壓下,維持其穩定的狀態,在這個情形下會有最小的體積改變,由以上三種因素,得以使分子動力學用數學式表現出來。
4.分散式運算
利用網路上的資料庫,來搜尋相關的資料,以拼湊出相關的結構﹔運用連接上網路的電腦,將其串聯起來,以發揮大型電腦所無法達到的運算能力,同時亦利用電腦進行藥物設計的模擬,而個人電腦使用window或是Linux的作業系統可以執行Folding@home 模擬程式,此一程式可以模擬蛋白質的結構。利用串聯的電腦方法的,是指先下載一個程式,而使用者平常可以正常的使用其電腦,只有在電腦閒置時,上述的程式會自動執行蛋白質的模擬,就好像美國太空總署(NASA)集合大家的力量,找尋外星智慧生命的例子一樣。
分散式運算實例:Snow et al., (2002) 針對一個小型蛋白質BBA5(約23個胺基酸具有較強的二級結構特性及斥水性核心),進行分子模擬與蛋白質生化實驗的摺疊比較,希望藉由電腦運算模擬出蛋白質快速摺疊的狀態,這需要極大量的運算,因此採用Folding@home分散式計算,將分布世界各地超過三萬名志願者的電腦組成一個大型叢集(cluster),花費數個月並累積約一百萬CPU天數的模擬時間,觀察到超過100種可能摺疊的方式,這是在一般系統所無法達成的模擬成果。
5.一維到三維
以現有的技術可以由蛋白質的一級結構預測三級結構,係利用已知的蛋白質結構對照目標蛋白質進行排序,找出目標蛋白質序列中二級結構的區域,並進一步的將其組合成三級結構。由胺基酸上的α-碳原子走向,可以順利的預測摺疊後骨架,但預測蛋白質結構則需參考β-碳原子,這是由於β-碳原子在螺旋(helix)或是摺板(sheet)中,能夠得到支鏈的相關訊息及環境。由序列對照到結構,首先採行穿針引線的方式,就是藉α-碳原子走向,將目標蛋白質的胺基酸由一級序列放置成三級結構,以完成蛋白質的骨架,之後再依實際的狀況,進一步的做修正。
6.生物資料庫
Entrez:由美國國家生物技術資料中心(NCBI, National Center for Biotechnology Information)所建置的網路資源,包含基因體資料庫及許多分析的工具(如BLAST, Basic Local Alignment Search Tool)。
Genbank:DNA序列資料庫,提供註冊、儲存及擷取DNA序列。
PDB:蛋白質結構資料庫,提供註冊、儲存及擷取蛋白質三維結構。
Swiss-Prot/TrEMBL:胺基酸序列資料庫,提供胺基酸序列註冊、註解及擷取。
7.蛋白質結構預測技術(CASP, Critical Assessment of technique for protein structure prediction)
這是在1994年所成立的實驗競賽,利用一個已知的蛋白質結構,由相關領域的研究人員,以不同的模擬技術來預測其結構,再與己知的結構做比較,來決定那種方式能最準確的預測出結構,並藉此用來評估模擬蛋白質結構方法的優缺點,目前已進行到CASP5。
CASP進行競賽的三個主要流程:
1.蒐集來自實驗社群的預測標的。
2.蒐集來自模擬社群的預測標的,並進行評估及討論結果。
3.將以上二種預測的標的,對照來自晶體結構及NMR結構的資訊。
晶體結構簡介
測定結構原理簡述:以X光測定分子結構的基本原理與顯微鏡類似。在一般光學或電子顯微鏡中,平行的輻射源(可見光或是電子束)通過切成薄片的樣品後,由於樣品本身質理密度分布不同,因此對入射的輻射源產生繞射的現象,置於樣品上方的光學物鏡,將繞射的光源補捉以後聚焦。及為實驗者在光學目鏡下所看到的放大影像。在實際操作上,X光繞射實驗與上述僅有些許的不同,目前沒有實質的光學透鏡能夠補捉聚焦的X光,因此必需經由光電轉換裝置或是照像顯影的方式,偵測紀錄晶體繞射X光,並呈現出對稱的幾何圖形,而此方法的巧妙之處,在於應用複雜的數學運算(傅立葉轉換)和高效率的電腦運算及圖像顯示系統,將繁瑣的X光繞射模型轉變成分子模型圖像。換句話說,在此方法中,我們所使用的是數學透鏡,而不是使用一般的光學透鏡。此方法所使用的樣品,也是大分子晶體學中的特色之一,如果比照顯微鏡放大的原理,實驗用的樣品,應該是巧妙的將一個生化分子固定在X光束中,然而在這種實驗情況下,分子繞射X光的強度將非常的微弱而無法偵測,因此使用結晶方法製備實驗樣品,將x光束通過樣品後,由於被測量的分子大幅度的增加,因此X光的強度也隨之增加。
結晶生化分子需要三種條件
1. 純度及數量
高純度的樣品容易形成單一數量均勻整齊排列的緊密個體。
2. 必須在適當的溶劑(一般為水-緩衝溶液)形成結晶
使用的緩衝液應與正常生理狀態下相同,以期實驗結果與實際不至相差很多。
3. 加入適量的沉澱劑,使溶液形成過飽和狀態
增加晶體形成與成長機會
目前最常使用的結晶法為蒸氣擴散實驗,將小量蛋白質等溶液加在玻璃載片上懸浮密封覆蓋在下層裝有較高濃度化學沉澱實驗劑的塑膠容器,密封靜置一段時間,由於上、下層溶液中沉澱劑濃度的差異,產生蒸發、擴散、對流的現象,因此將沉澱劑慢慢的滲入上層珠狀生化分子溶液中,形成飽和穩定狀態,降低其溶解度而後形成晶種,逐漸形成單晶樣品。影響結晶的因素有:生化分子的純度、溫度、酸鹼值、振動與聲音、金屬離子緩衝溶液的成份、結晶的體積及晶種、沉澱劑的濃度。
8.NMR
基本原理:核磁共振波譜學是探討物質與電磁波的作用,屬於光譜學的一支。物質如何受到影響,仰賴於射頻波的波長與頻率,科學家利用所得結果了解有關物質在分子層次的問題,探討原子核磁特性所表現出的行為。核磁共振的原理如下:物質中的原子核,其行為就有如微小的指南針般,稱為核自旋,其行為就好像微小的指南針一樣。將物質放在靜磁場中,每個小指南針和磁場所成的角度相當於核自旋處於不同的時期,由於有溫度效應,使得低能階的分布略多。當射頻波的頻率剛好等於兩能階間的差異,低能階的核自旋會遷升到高能階,此即核磁共振。為測不同能階的差異,可緩慢掃描射頻頻率,當共振時電磁波被吸收,偵測線路阻抗改變,引發電子訊號把訊號強度對頻率作圖,所得圖譜稱為核磁共振波譜,而核磁共振的差異不僅和原子核的自身性質有關,也跟所處化學環境有關,因此核磁共振開啟了在化學的應用。核磁共振的訊號,亦可用來決定化合物中的分子結構,在早期,由於核磁共振方法靈敏相當低,他的應用僅限於研究濃度高的分子,而近代改用短而強的射頻脈衝取代掃描頻率方法提高靈敏度。脈衝和自旋作用產生訊號,此訊號隨隨時間遞減,直接觀察對時間變化的訊號,不易了解其意義,經過傅立葉方程式轉換以後,可得一般的波譜。而且靈敏度的提高可由訊號累加而得,累加所用的時間較傳統掃描方法大大的減少。核磁共振波譜是指譜線強度對頻率的變化,所以稱這種光譜為一維光譜。
注釋:
1、Protein Structure Resource:http://psr.life.nthu.edu.tw/
2、生物資訊群體學習網站:http://elearning.bioinfo.ntu.edu.tw/
3、http://psr.life.nthu.edu.tw/psm/hm.htm
4、Nature Structural Biology 7, 986 - 990 (2000)
5、Nature Structural Biology 7, 986 - 990 (2000)
6、Stockholm Bioinformatics Center:http://www.sbc.su.se/
7、輔仁大學生命技術研發中心:http://brc.se.fju.edu.tw/ (黃明經博士演講投影片)
8、生物資訊群體學習網站:http://elearning.bioinfo.ntu.edu.tw/
9.參考資料:
http://www.shmu.edu.cn/cc/11.doc
http://mod.life.nthu.edu.tw/homology/teachenglish.htm
http://www.ncku.edu.tw/~cbst/Oldhomepage/bioinfostructure.htm
http://www.chemcomp.com/feature/bio1999.htm#Search
Application to chemistery and chemical physics
http://predictioncenter.llnl.gov/
http://www.salilab.org/~andras/watanabe/node11.html
http://www.biocomp.unibo.it/school/html2003/ABSTRACT/bystroff.html
http://isites.bio.rpi.edu/Isites/
http://depts.washington.edu/bakerpg/abstracts/Bystroff_HMMSTR_JMB2000.html
http://www.ijc.com/abstracts/keywords.html
http://www.aiche.org/conferences/techprogram/paperdetail.asp?PaperID=1720&DSN=annual01
http://fulcrum.physbio.mssm.edu:8083/research.html.
http://www.sbc.su.se/~maccallr/teaching/SU/D-course/Lecture-two.pdf
http://www.wspc.com/books/physics/4774.html
http://www.uni-tuebingen.de/uni/opx/reports/roth_111.ps.gz
http://homepages.nyu.edu/~mt33/PINPT/node1.html
http://homepages.nyu.edu/~mt33/PINY_MD/PINY.html