ExPASy資料庫簡介-蛋白質序列分析與預測(1)
口述:呂平江 老師
整理:林崇文、鄭詩思、李佩真
1.Protein identification and characterization
4.Pattern and profile searches
5.Post-translational modification prediction
8.Secondary structure prediction

1.
Protein identification and characterization
蛋白質鑑定及特性描述
2.
DNA -> Protein
將DNA序列轉為蛋白質序列的工具
3.
Similarity searches
相似序列的搜尋
4.
Pattern and profile searches
模型及圖表的搜尋
5.
Post-translational modification prediction
預測轉譯時的修飾
6.
Topology prediction
拓樸學的預測
7.
Primary structure analysis
主要結構的分析
8.
Secondary structure prediction
次級結構預估
9.
Tertiary structure
三級結構
10.
Sequence alignment
序列比對
11.
Biological text analysis
生物學主題分析

1.Protein identification and characterization
蛋白質鑑定及特性描述
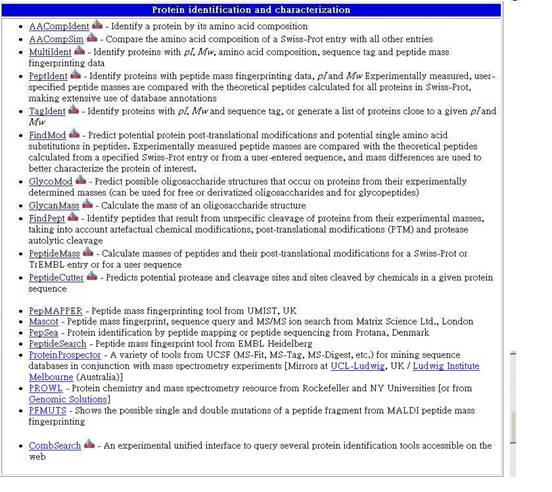
1-1 AACompIdent - Identify a protein by its amino acid composition
以胺基酸成分鑑定蛋白質。
1-2 AACompSim - Compare the amino acid composition of a Swiss-Prot entry with all other entries
比較Swiss-Prot entry 與其他entries 胺基酸成分分析結果的差異。
1-3 MultiIdent - Identify proteins with pI, Mw, amino acid composition, sequence tag and peptide mass fingerprinting data
以等電點、分子量、胺基酸成分、序列尾端及胜肽質譜特徵資料鑑定蛋白質。
1-4 PeptIdent - Identify proteins with peptide mass fingerprinting data, pI and Mw Experimentally measured, user-specified peptide masses are compared with the theoretical peptides calculated for all proteins in Swiss-Prot, making extensive use of database annotations
以胜肽質譜特徵資料、等電點、分子量、胺基酸成分、序列尾端來鑑定蛋白質,以Swiss-Prot中所有蛋白質的理論性胜肽來比較實驗上的測量以及使用者指定的胜肽質譜,提供廣泛的資料庫註解功能。
1-5 TagIdent - Identify proteins with pI, Mw and sequence tag, or generate a list of proteins close to a given pI and Mw
以等電點、分子量、序列尾端來鑑定蛋白質,並產生與所給之等電點及分子量最接近的蛋白質列表。
1-6 FindMod - Predict potential protein post-translational modifications and potential single amino acid substitutions in peptides. Experimentally measured peptide masses are compared with the theoretical peptides calculated from a specified Swiss-Prot entry or from a user-entered sequence, and mass differences are used to better characterize the protein of interest.
預測可能的蛋白質轉譯修飾及單一胺基酸可能取代之胜肽。將實驗上的胜肽質譜測量結果與指定的Swiss-Prot中的理論性胜肽或使用者所輸入的序列作比較,集合其差異以作出更佳的蛋白質特性描述。
1-7 GlycoMod - Predict possible oligosaccharide structures that occur on proteins from their experimentally determined masses (can be used for free or derivatized oligosaccharides and for glycopeptides)
以實驗結果的質譜預測可能發生在蛋白質中的低級多醣結構。〈可免費使用並由糖蛋白中的低級多醣來加以引導〉
1-8 GlycanMass - Calculate the mass of an oligosaccharide structure
由一低級多醣蛋白的結構預測其質譜。
1-9 FindPept - Identify peptides that result from unspecific cleavage of proteins from their experimental masses, taking into account artefactual chemical modifications, post-translational modifications (PTM) and protease autolytic cleavage
鑑定一由實驗質譜結果所得的蛋白質胜肽,並考慮到人工的化學修飾、轉譯修飾(PTM)及蛋白質自體溶解等因素。
1-10 PeptideMass - Calculate masses of peptides and their post-translational modifications for a Swiss-Prot or TrEMBL entry or for a user sequence
以Swiss-Prot 、TrEMBL entry或使用者所提供的序列來預測其胜肽質譜及轉譯時的修飾。
1-11 PeptideCutter - Predicts potential protease and cleavage sites and sites cleaved by chemicals in a given protein sequence
由所提供的蛋白質序列來預測可能的蛋白質及其化學切開點。
1-12 PepMAPPER - Peptide mass fingerprinting tool from UMIST, UK
由UMIST及 UK所提供的胜肽質譜鑑定工具。
1-13 Mascot - Peptide mass fingerprint, sequence query and MS/MS ion search from Matrix Science Ltd., London
到Matrix Science Ltd., London搜尋MS/MS離子及進行胜肽質譜鑑定。
1-14 PepSea - Protein identification by peptide mapping or peptide sequencing from Protana, Denmark
以胜肽繪圖及胜肽排序來鑑定蛋白質,由Protana, Denmark提供。
1-15 PeptideSearch - Peptide mass fingerprint tool from EMBL Heidelberg
由EMBL Heidelberg所提供的胜肽質譜鑑定工具。
1-16 ProteinProspector - A variety of tools from UCSF (MS-Fit, MS-Tag, MS-Digest, etc.) for mining sequence databases in conjunction with mass spectrometry experiments [Mirrors at UCL-Ludwig, UK / Ludwig Institute Melbourne (Australia)]
一個由UCSF所提供的多樣性質譜分析工具。
1-17 PROWL - Protein chemistry and mass spectrometry resource from Rockefeller and NY Universities [or from Genomic Solutions]
由Rockefeller and NY Universities提供蛋白質化學性質及質譜儀資源。
1-18 PFMUTS - Shows the possible single and double mutations of a peptide fragment from MALDI peptide mass fingerprinting
由MALDI提供,呈現一胜肽質譜鑑定及胜肽碎片可能的單一或雙重變化。
1-19 CombSearch - An experimental unified interface to query several protein identification tools accessible on the web
一是試驗性的蛋白質鑑定工具連結介面,相當容易在網上進入。
將DNA序列轉為蛋白質序列的工具

2-1 Translate - Translates a nucleotide sequence to a protein sequence
將一DNA序列轉為蛋白質序列。
2-2 Transeq - Nucleotide to protein translation from the EMBOSS package
使用EMBOSS package將核甘酸轉為蛋白質
2-3 Graphical Codon Usage Analyser - Displays the codon bias in a graphical manner
以寫實方法呈現密碼子偏差。
2-4 BCM search launcher - Six frame translation of nucleotide sequence(s)
核甘酸序列的六種翻譯結構。
2-5 Backtranslation - Translates a protein sequence back to a nucleotide sequence
將一蛋白質序列翻譯回核甘酸序列。
2-6 Genewise - Compares a protein sequence to a genomic DNA sequence, allowing for introns and frameshifting errors
將蛋白質的序列與基因組的DNA序列做比較,並考慮到intron及frameshifting的錯誤。
2-7 FSED - Frameshift error detection
偵測frameshifting的錯誤。
2-8 LabOnWeb - Elongation, expression profiles and sequence analysis of ESTs using Compugen LEADS clusters
使用Compugen LEADS clusters做延伸、展現圖表及ESTs的序列分析。
2-9 List of gene identification software sites
列出基因鑑定的軟體。
相似序列的搜尋

3-1 BLAST and WU-BLAST - Interfaces to various versions of the Basic Local Alignment Search Tool
Basic Local Alignment Search 工具的不同翻譯連結。
3-2 BLAST Network Service on ExPASy
ExPASy 的BLAST Network伺服器。
3-3 BLAST at EMBnet-CH/SIB (Switzerland)
EMBnet-CH/SIB (Switzerland) 的BLAST。
3-4 BLAST at NCBI
NCBI的BLAST。
3-5 WU-BLAST at Bork's group in EMBL (Heidelberg)
EMBL (Heidelberg) Bork's group的WU-BLAST。
3-6 WU-BLAST and BLAST at the EBI (Hinxton)
EBI (Hinxton) 的WU-BLAST及BLAST。
3-7 BLAST at PBIL (Lyon)
PBIL (Lyon) 的BLAST。
3-8 Bic ultra-fast rigorous (Smith/Waterman) similarity searches using the Bioccelerator [At DKFZ or at Weizmann]
使用Bioccelerator的Bic ultra-fast rigorous (Smith/Waterman)來精確搜尋相似的序列。
3-9 MPsrch - Smith/Waterman sequence comparison at EBI
位於EBI的Smith/Waterman序列比對。
3-10 DeCypher - Smith/Waterman or FrameSearch search using the DeCypher hardware accelerator
使用DeCypher hardware accelerator的Smith/Waterman 或 FrameSearch序列搜尋。
3-11 Fasta3 - FASTA version 3 at the EBI
位於EBI 的FASTA version 3
3-12 FDF - Smith/Waterman type searches on Paracel's Fast Data Finder (FDF) at EMBnet-CH
位於EMBnet-CH 中Paracel's Fast Data Finder (FDF)上的Smith/Waterman 式搜尋。
3-13 PropSearch; searches for structural homologs using a 'properties' approach [At EMBL or at Montpellier]
使用EMBL 或 Montpellier搜尋相近的結構。
3-14 SAMBA - Systolic Accelerator for Molecular Biological Applications
心縮加速裝置在分子生物學上的應用。
3-15 SAWTED - Structure Assignment With Text Description
描述結構功能。
3-16 Scanps - Similarity searches using Barton's algorithm
使用Barton's algorithm搜尋相似序列。
4.Pattern and profile searches
模型及圖表的搜尋

4-1 InterPro Scan - Integrated search in PROSITE, Pfam, PRINTS and other family and domain databases
在PROSITE, Pfam, PRINTS及其他家族資料庫中合併搜尋。
4-2 ScanProsite - Scans a sequence against PROSITE or a pattern against Swiss-Prot and TrEMBL
掃描一段相對於PROSITE、Swiss-Prot 和TrEMBL的序列及圖表。
4-3 MotifScan - Scans a sequence against protein profile databases (including PROSITE)
掃描一段相對於protein profile資料庫的序列(包括PROSITE)。
4-4 Frame-ProfileScan - Scans a short DNA sequence against protein profile databases (including PROSITE)
掃描一段相對於protein profile資料庫的短鏈DNA(包括PROSITE)。
4-5 Pfam HMM search; scans a sequence against the Pfam protein families db [At Washington University or at Sanger Centre]
在Washington University及Sanger Centre掃描一段相對於Pfam protein families db的序列(包括PROSITE)。
4-6 FingerPRINTScan - Scans a protein sequence against the PRINTS Protein Fingerprint Database
掃描一段相對於PRINTS Protein Fingerprint資料庫的蛋白質序列。
4-7 FPAT - Regular expression searches in protein databases
蛋白質資料庫中的規律表現搜尋。
4-8 PRATT - Interactively generates conserved patterns from a series of unaligned proteins; [at EBI / ExPASy ]
在EBI 及ExPASy產生一unaligned蛋白質的圖表。
4-9 PPSEARCH - Scans a sequence against PROSITE (allows a graphical output); at EBI
在EBI中掃描一段相對於PROSITE的序列(允許產生圖解)。
4-10 PROSITE scan - Scans a sequence against PROSITE (allows mismatches); at PBIL
在PBIL掃描一段相對於PROSITE的序列(允許錯誤)。
4-11 PATTINPROT - Scans a protein sequence or a protein database for one or several pattern(s); at PBIL
在PBIL掃描一段蛋白質序列或蛋白質資料庫的圖表。
4-12 SMART - Simple Modular Architecture Research Tool; at EMBL
位於EMBL的一個簡單的分子結構搜尋工具。
4-13 TEIRESIAS - Generate patterns from a collection of unaligned protein or DNA sequences; at IBM
於IBM產生一unaligned蛋白質或DNA序列的圖表。
4-14 Hits - Relationships between protein sequences and motifs
蛋白質結構與motifs間的關係。
5.Post-translational modification prediction
預測轉譯時的修飾

5-1 SignalP - Prediction of signal peptide cleavage sites
預測信號胜 的切點。
5-2 ChloroP - Prediction of chloroplast transit peptides
葉綠體傳送胜肽的預測。
5-3 MITOPROT - Prediction of mitochondrial targeting sequences
預測粒腺體的目標序列。
5-4 Predotar - Prediction of mitochondrial and plastid targeting sequences
預測粒腺體與質體的目標序列。
5-5 NetOGlyc - Prediction of O-GalNAc (mucin type) glycosylation sites in mammalian proteins
預測哺乳動物體內黏蛋白型態的蛋白質醣化位置。
5-6 NetNGlyc - Prediction of N-glycosylation sites in human proteins
預測人類體內的N型蛋白質醣化位置。
5-7 DictyOGlyc - Prediction of GlcNAc O-glycosylation sites in Dictyostelium
預測黏菌體內的O型蛋白質醣化位置。
5-8 YinOYang - O-beta-GlcNAc attachment sites in eukaryotic protein sequences
真核生物蛋白質序列上O-beta-GlcNAc的附著位置。
5-9 big-PI Predictor - GPI Modification Site Prediction
預測GPI的修飾位置。
5-10 DGPI - Prediction of GPI-anchor and cleavage sites (Mirror site)
預測GPI的切點與接點位置(鏡像站)。
5-11 NetPhos - Prediction of Ser, Thr and Tyr phosphorylation sites in eukaryotic proteins
真核生物蛋白質上Ser, Thr 及 Tyr phosphorylation位置的預測。
5-12 NetPicoRNA - Prediction of protease cleavage sites in picornaviral proteins
picornaviral proteins上切點的預測。
5-13 NMT - Prediction of N-terminal N-myristoylation
N-terminal N-myristoylation的預測。
5-14 Sulfinator - Prediction of tyrosine sulfation sites
酪胺酸硫化位置的預測。
5-15 SUMOplot - Prediction of SUMO protein attachment sites
SUMO蛋白質附著位置的預測。
拓樸學的預測

6-1 PSORT - Prediction of protein sorting signals and localization sites
預測蛋白質的sorting 信號及localization sites。
6-2 TargetP - Prediction of subcellular location
預測次細胞的位置。
6-3 DAS - Prediction of transmembrane regions in prokaryotes using the Dense Alignment Surface method (Stockholm University)
使用Dense Alignment Surface法預測原核生物的transmembrane區域。
6-4 HMMTOP - Prediction of transmembrane helices and topology of proteins (Hungarian Academy of Sciences)
預測蛋白質的transmembrane helices (跨膜螺旋)及 拓樸學。
6-5 PredictProtein - Prediction of transmembrane helix location and topology (Columbia University)
預測蛋白質的transmembrane helices (跨膜螺旋)及 拓樸學。
6-6 SOSUI - Prediction of transmembrane regions (TUAT; Tokyo Univ. of Agriculture & Technology)
預測transmembrane區域。
6-7 TMAP - Transmembrane detection based on multiple sequence alignment (Karolinska Institut; Sweden)
以複合序列為基礎偵測Transmembrane。
6-8 TMHMM - Prediction of transmembrane helices in proteins (CBS; Denmark)
預測蛋白質的transmembrane helices (跨膜螺旋)
6-9 TMpred - Prediction of transmembrane regions and protein orientation (EMBnet-CH)
預測transmembrane區域及蛋白質方位。
6-10 TopPred 2 - Topology prediction of membrane proteins (Stockholm University)
膜蛋白的拓樸學預測。
主要結構的分析

7-1 ProtParam - Physico-chemical parameters of a protein sequence (amino-acid and atomic compositions, pI, extinction coefficient, etc.)
蛋白質序列的物理及化學變量分析(胺基酸、原子結構、等電點….等)
7-2 Compute pI/Mw - Compute the theoretical pI and Mw from a Swiss-Prot or TrEMBL entry or for a user sequence
以Swiss-Prot or TrEMBL entry 或使用者提供序列估算其等電點及分子量。
7-3 MW, pI, Titration curve - Computes pI, composition and allows to see a titration curve
估算等電點及成分並可看見其滴定曲線圖。
7-4 REP - Searches a protein sequence for repeats
搜尋重複的蛋白質序列。
7-5 REPRO - De novo repeat detection in protein sequences
重新偵測重複的蛋白質序列。
7-6 Radar - De novo repeat detection in protein sequences
重新偵測重複的蛋白質序列。
7-7 SAPS - Statistical analysis of protein sequences at EMBnet-CH [Also available at EBI]
EMBnet-CH及EBI的蛋白質序列統計分析。
7-8 Coils - Prediction of coiled coil regions in proteins (Lupas's method) at EMBnet-CH [Also available at PBIL]
於EMBnet-CH 及 PBIL預測蛋白質的卷曲螺旋區域。
7-9 Paircoil - Prediction of coiled coil regions in proteins (Berger's method)
以Berger's法預測蛋白質的卷曲螺旋區域。
7-10 Multicoil - Prediction of two- and three-stranded coiled coils
預測蛋白質二、三級結構的卷曲螺旋。
7-11 2ZIP - Prediction of Leucine Zippers
亮氨酸拉鍊的預測。
7-12 PEST - Identification of PEST regions
PEST區域的鑑定。
7-13 PESTfind - Identification of PEST regions at EMBnet Austria
以EMBnet Austria 鑑定PEST的區域。
7-14 HLA_Bind - Prediction of MHC type I (HLA) peptide binding
預測MHC type I (HLA) 的peptide binding。
7-15 SYFPEITHI - Prediction of MHC type I and II peptide binding
預測MHC type I 及 II的 peptide binding。
7-16 ProtScale - Amino acid scale representation (Hydrophobicity, other conformational parameters, etc.)
胺基酸刻度圖(構造及其他參數等)。
7-17 Drawhca - Draw an HCA (Hydrophobic Cluster Analysis) plot of a protein sequence
以Hydrophobic Cluster Analysis繪出蛋白質序列。
7-18 Protein Colourer - Tool for coloring your amino acid sequence
將您的胺基酸序列上色的工具。
7-19 Three To One - Tool to convert a three-letter coded amino acid sequence to single letter code
將三碼的胺基酸序列轉為單一密碼的工具。
7-20 Colorseq - Tool to highlight (in red) a selected set of residues in a protein sequence
將所選擇的蛋白質序列以紅色加以強調的工具。
7-21 HelixWheel / HelixDraw - Representations of a protein fragment as a helical wheel
蛋白質片段的環狀螺旋結構模型。
7-22 RandSeq - Random protein sequence generator
隨機的蛋白質序列產生器。
8.Secondary structure prediction
次級結構預估

8-1 AGADIR - An algorithm to predict the helical content of peptides
一個預估胜肽鏈螺旋結構的演算法。
8-2 BCM PSSP - Baylor College of Medicine
Baylor藥物學院。
8-3 Prof - Cascaded Multiple Classifiers for Secondary Structure Prediction
以Cascaded Multiple Classifiers進行次級結構預估。
8-4 GOR I (Garnier et al, 1978) [At PBIL or at SBDS]
GOR I次級結構預估法
8-5 GOR II (Gibrat et al, 1987)
GOR II次級結構預估法
8-6 GOR IV (Garnier et al, 1996)
GOR IV次級結構預估法
8-7 HNN - Hierarchical Neural Network method (Guermeur, 1997)
Hierarchical Neural Network次級結構預估法
8-8 Jpred - A consensus method for protein secondary structure prediction at University of Dundee
Dundee大學提供的一般性次級結構預估法。
8-9 nnPredict - University of California at San Francisco (UCSF)
San Francisco的California大學。
8-10 PredictProtein - PHDsec, PHDacc, PHDhtm, PHDtopology, PHDthreader, MaxHom, EvalSec from Columbia University
Columbia大學的PHDsec, PHDacc, PHDhtm, PHDtopology, PHDthreader, MaxHom, EvalSec
8-11 PSA - BioMolecular Engineering Research Center (BMERC) / Boston
波士頓分子生物學研究中心。
8-12 PSIpred - Various protein structure prediction methods at Brunel University
Brunel大學所提供之多種蛋白質結構預估法。
8-13 SOPM (Geourjon and Del嶧ge, 1994)
SOPM次級結構預估法。
8-14 SOPMA (Geourjon and Del嶧ge, 1995)
SOPMA次級結構預估法。
三級結構

9-1 SWISS-MODEL - An automated knowledge-based protein modelling server
能自動化產生蛋白質模型的伺服器。
9-2 Geno3d - Automatic modelling of protein three-dimensional structure
可自動產生蛋白質3D立體結構模型。
9-3 CPHmodels - Automated neural-network based protein modelling server
能自動化產生蛋白質模型的伺服器。
9-4 3D-PSSM - Protein fold recognition using 1D and 3D sequence profiles coupled with secondary structure information (Foldfit)
使用次要架構資訊結合的蛋白質摺疊 1D和 3D的序列資料的識別 ( Foldfit ) 。
9-5 ProSup - Protein structure superimposition
蛋白質結構的疊合。
9-6 SWEET - Constructing 3D models of saccharids from their sequences
依照序列建立糖類的3D結構模型。
9-7 Swiss-PdbViewer - A program to display, analyse and superimpose protein 3D structures
可將蛋白質3D結構摺疊、分析並展示的程式。
序列比對

10-1 Binary (雙重)
10-1-1 SIM + LALNVIEW - Alignment of two protein sequences with SIM, results can be viewed with LALNVIEW
以SIM比對二序列,其結果可以LALNVIEW觀看。
10-1-2 LALIGN - Finds multiple matching subsegments in two sequences
找到二序列多個相同的部分。
10-1-3 Dotlet - A Java applet for sequence comparisons using the dot matrix method
使用Java applet 呈現以dot matrix method比較序列的結果。
10-2 Multiple (多重)
10-2-1 CLUSTALW [At EBI, PBIL or at EMBnet-CH]
以EBI, PBIL或EMBnet-CH比對序列。
10-2-2 T-Coffee [At EMBnet Switzerland or at CMBI]
以EMBnet Switzerland或CMBI比對序列。
10-2-3 ALIGN - at Genestream (IGH)
以Genestream (IGH)比對序列。
10-2-4 DIALIGN - Multiple sequence alignment based on segment-to-segment comparison, at University of Bielefeld, Germany
德國Bielefeld大學提供的複合序列比對。
10-2-5 Match-Box - at University of Namur, Belgium
比利時Namur大學提供的複合序列比對。
10-2-6 MSA - at Washington University
Washington大學提供的複合序列比對。
10-2-7 Multalin [At INRA or at PBIL]
在INRA或 PBIL比對複數序列。
10-2-8 MUSCA - Multiple sequence alignment using pattern discovery, at IBM
IBM 提供的pattern discovery複合序列比對。
10-2-9 AMAS - Analyse Multiply Aligned Sequences
用來分析複合序列。
10-2-10 Bork's alignment tools - Various tools to enhance the results of multiple alignments (including consensus building).
提升一些聯合結果 (包括一致結果) 的各種工具。
10-2-11 CINEMA - Color Interactive Editor for Multiple Alignments
為分析結果著色的工具。
10-2-12 ESPript - Tool to print a multiple alignment
複合序列比對結果列印工具。
10-2-13 plogo - Sequence logos at CBS/Denmark
丹麥CBS的Sequence logos。
10-2-14 GENIO/logo - Sequence logos at Stuttgart/Germany
德國Stuttgart的Sequence logos。
10-2-15 WebLogo - Sequence logos at Cambridge/UK
英國劍橋的Sequence logos。
生物學主題分析

11-1 AcroMed - A computer generated database of biomedical acronyms and the associated long forms extracted from the recent Medline abstracts
生物醫學縮寫及結合字的電子資料庫。
11-2 AbXtract - A prototype system for the automatic annotation of functional characteristics in protein families
蛋白質家族功能性特徵自動註解系統的原型。
11-3 MedMiner - Extract and organize relevant sentences in the literature based on a gene, gene-gene or gene-drug query
提供有關gene, gene-gene 或 gene-drug的查詢。
11-4 Protein Annotator's Assistant - A software system which assists protein annotators in the task of assigning functions to newly sequenced proteins
一個協助蛋白質註解的軟體,可將最新的蛋白質序列加以分配功能。
11-5 XplorMed - Explore a set of abstracts derived from a bibliographic search in MEDLINE
由MEDLINE搜尋相關資訊。