蛋白質結構資料庫介紹Structure Database
整理:李佩曄、許秀惠、郭昱辰
2.The Notion of Three-Dimensional Molecular Structure Data
3.Coordinates、Sequence、and chemical Graphs
4.Atoms、Bonds、and Completeness
8.Sequence from Structure Records
10.MMDB:MOLECULAR MODELING DATABASE AT NCBI
11.Free Text Query of Structure Records
13.BLAST Against PDB Sequences: New Sequences Similarities
14.Entrez Neighboring: Known Sequence Similarities
15.Visualizing structural information
16.Picture the Data: Populations, Degeneracy, and Dynamics
21.RasMol and RasMol-based Viewers
23.Other 3D Viewers:Mage、CAD、and VRML
24.Making Presentation Graphics
25.Advanced structure modeling
26.Structure similarity searching
28.Internet resources for topic presented
1.Introduction to structure
這章介紹生物分子結構是從藉由強調3D結構序列的生物資訊perspective(透視畫法)得知,這章的主要目的是向讀者報告結構database的內容,以及如何處理或有時由軟體去處理此資料。
蛋白質和核酸結構的影像變成生化教科書和研究文章的普遍特徵,而這個影像主要可由X光晶體繞射或者是由nuclear magnetic resonance (NMR) spectroscopists,而這些結構資訊可能提供了關於蛋白質功能重要的資訊。而對此Protein Data Band(PDB)從Brookhaven National Laboratories 移到Research Collaboratory for structure Biology。此將對生物學的改變造成衝擊。
2.The Notion of Three-Dimensional Molecular Structure Data
讓我們開始一個智力的訓練在於一個biopolymer之Three-Dimensional結構 Data的紀錄,例如myoglobin 詳細的Three-Dimensional ball-and-stick model。我們可藉由其序列得到Three-Dimensional model 的backbone,從N-端開始由和20個胺基酸的化學結構比較其每個residue atomic structure。
一旦將序列寫下,我們可製出biopolymer所有原子的 Two-Dimensional sketch、element symbols、and Bond可能佔有的部分,在畫出他的化學Three-Dimensional結構之後,我們可以紀錄Three-Dimensional data藉由orthogonal axis system 從一些原點到每一個原子的距離,這將可以提供x-,y-,and z-axis到每個原子的距離在ball-and-stick model structure,而下個步驟則是使所有的(x, y and z)協調的連結去定義每個原子。
而這個實驗使我們對於Three-Dimensional structure database 的紀錄更有概念,有兩件事是我們必須注意的,每個原子在空間中的1.化學結構及2.位置。
3.Coordinates、Sequence、and chemical Graphs
Coordinates data 為分子原子在空間中座落的位置,這些資料是由x,y,z沿著空間中每一條axis來呈現的,我們可以直接從sequence推斷biopolymer molecular 的化學性連接,包含其原子及鍵結,並且也可以利用sequence的資訊來製作Three-Dimensional structure 的chemical graph。
當我們根據sequence所呈現的Three-Dimensional、原子、鍵結來製圖時,通常要遵從textbook顯示的residue之化學結構,以避免忘了methyl group,而且電腦建立的化學結構圖形會以residue dictionary 為基準,且這包含了胺基酸及核酸的原子及鍵結資訊。
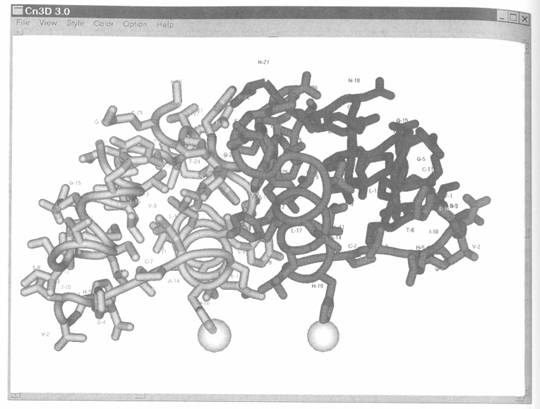
Fig.1. The insulin structure
4.Atoms、Bonds、and Completeness
Molecular graphics visualization software 實行精確的connect-the-dots過程製造蛋白質結構的圖形,如圖1中Insulin的結構圖。
研究原子和鍵結的紀錄有時我們叫做chemistry rules approach,這個規則是一種化學的物理規則觀察,例如平均穩固的C-C鍵之間的長度為1.5 angstroms。利用這個規則便可以個別的C原子之間的single bond 為1.5 angstroms。因此我們便可以忽略bond,使一個完美且完整的結構可以被紀錄且不需鍵結的資料。而PDB的格式是缺少biopolymer 的鍵結資訊,因此在PDB檔案中的bounding是交給程式去決定,而且結果在他所畫的鍵結會是不一致的,特別是在不同algorithms。
Explicit bonding approach,此方法是使用在Molecular Modeling Database(MMDB)的Database的紀錄,在Molecular Modeling Database(MMDB)系統the data是包含了explicit bonding approach 的information,Molecular Modeling Database(MMDB)利用標準的residue dictionary ,一個記錄所有的原子及鍵結在氨基酸及核酸residue的polymer forms中,而這通常利用到X-ray or NMR structure,在Molecular Modeling Database(MMDB)的軟體中可以利用bonding information 提供一個方向去連接原子而不需利用chemistry rule。
科學家通常會以database去預期結構,但常常驚訝其中的差異,主要是因為沒有符合結構的completeness,因為:A least one coordinate value for each and every atom in the chemical graph is present。
PDB: PROTEIN DATA BANK AT THE RESEARCH COLLABORATORY FOR STRUCTURAL BIOINFORMATIC(RCSB)
5.Overview
電腦在生物學上的使用起源於生物物理的方法,例如:X-ray crystallography及NMR spectroscopists ,而這些收集的 bioinformation database則由RESEARCH COLLABORATORY FOR STRUCTURAL BIOINFORMATIC(RCSB)經營的PDB存取。
6.RCSB Database Services
在RCSB的Protein database bank提供了糾正其Three-Dimensional structure data 上的服務,也提供一些Three-Dimensional的資料及相關軟體在其網頁上,讓使用者下載。
7.PDB Query and reporting
從RCSB的網頁我們可以得到Three-Dimensional structure 藉由使用兩種不同的引擎,而SearchLite system是最常使用的,可經由database提供主題收尋,如圖2。

Fig.2. Structure query from RCSB with the structure 1BNR.
Submitting Structures. 為了使Three-Dimensional structure information 符合PDB,RCSB 提供他的ADIT service在網頁上,這個service 提供了data format check 並且可以診斷結構,如鍵結的距離、鍵結角度、扭轉的角度、nucleic acid comparison and crystal packing。
PDB-ID Codes.PDB的結構記錄以一個獨一的four-character alphanumeric code,稱之為PDB-ID or PDB code,此方法使用了數字0-9及單字A-Z,這便超過了1.3 million 的組合。
Database searching , PDB File Retrieval , mmCIF File Retrieval, and Links.PDB的收尋引擎,the structure Explorer可以reyrieve PDB records,如圖2表示出structure summary for the protein barnase,The Structure Explorer 也提供了database相關主題的連結,如structure-structure similarity,and protein motion。
8.Sequence from Structure Records
Explicit sequence:
在PDB file以關鍵字SEQRES被提供,不像其他sequence database,PDB 的紀錄以three-letter amino acid code。但這似乎太明確,因為雙股DNA的5-端到3-端及另一股3-端到5-端對使用者來說是可以觀察到的,但對電腦而言3-端到5-端卻是nonsense。
Implicit sequence:
此implicit sequence 在PDB 記錄裡是包含(x,y,z)data的stereochemistry,以及在PDB file 的ATOM record,implicit sequence可解決explicit sequence ambiguities如backward encoding of nucleic acid sequence or nonstandard 氨基酸的檢驗。如圖3。
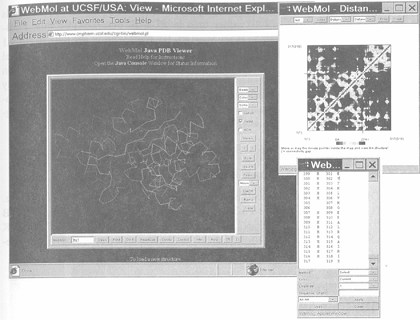
Fig.3.
Testing a three dimesional viewer for sequence numbering artifacts with
the structure 3TS1
※參考資料:Bioinformatics
在做功能分析時,若找到一個重要的蛋白質時,通常需要知道此蛋白質的結構,以瞭解其作用機制,甚至用電腦軟體設計可能和此蛋白質結合,而影響其功能的小分子。因此需要查閱資料庫中是否有其結構資訊,若沒有結構資訊,則需檢視要研究的蛋白質是否與已知結構的蛋白質具同源性。如果具有同源性,則可用利用軟體由已知的結構做模擬,來預測所要研究的蛋白質之可能結構。
主要的巨分子結構資料庫有兩個,包括以收集蛋白質結構為主的PDB (Protein Data Bank),和以收集核酸結構為主的NDB (The Nucleie Acid DataBase)。
PDB 除了收錄蛋白質結構外,亦有核酸結構與少數分子模擬之結構。NDB 中大部份的結構均來自於 PDB,因此多具有 PDB 的代碼,只有少數核酸結構是直接存入 NDB,而無 PDB 代碼。
◎資料庫
|
資料庫形式 |
資料庫名稱 |
格式 |
評 論 |
|
原始數值資料庫 |
NDB (Nucleic acid database) |
|
核酸結構資料庫 |
|
|
PDB (Protein database) |
蛋白質結構資料庫 |
|
|
加值資料庫 |
MMDB (Molecular modelling database) |
asn.1 |
|
|
|
|
以各種不同之格式儲存分子模型 |
|
|
|
|
可連接至蛋白質序列,結構分類,性質,與突變等多個資料庫 |
9.Validating PDB Sequences
在美國生物技術資料中心的MMDB(Molecular Modeling DataBase),是用其自己的方式整理 PDB 的資料,所以查詢MMDB與PDB會得到同樣的資訊,因此MMDB是確定sequence的最好方法。
10.MMDB:MOLECULAR MODELING DATABASE AT NCBI
查詢MMDB及PDB資料庫所用的工具不同,查詢前者是用「Entrez」,而查詢後者是使用「3DB Browser」。在Entrez 的查詢結果中,不但可連到 MMDB,更可連到序列與文獻的資訊,對於更專業領域問題作者推薦RCSB;3DB Browser 雖然也做的不錯,卻不如 Entrez 好用。而MMDB的格式是ASN.1。
11.Free Text Query of Structure Records
MMDB database可以利用Entrez方式來搜尋,若是需要更詳細的資料則推薦RCSB這個網站。
12.MMDB Structure Summary
MMDB 中除了可以將結構下載成為 Cn3D,MAGE 與 PDB 等三種格式外,更可連到「相關的」序列或結構,這是 PDB 檔案中沒有的連接。相關的序列雖然很容易找,可是結構上的相似性,卻不好找。在 MMDB 中是利用向量並列分析 (Vector alignment Search Tool;VAST) 的方法做的,其結果比序列上的相似性更容易推斷蛋白質間的同源性。
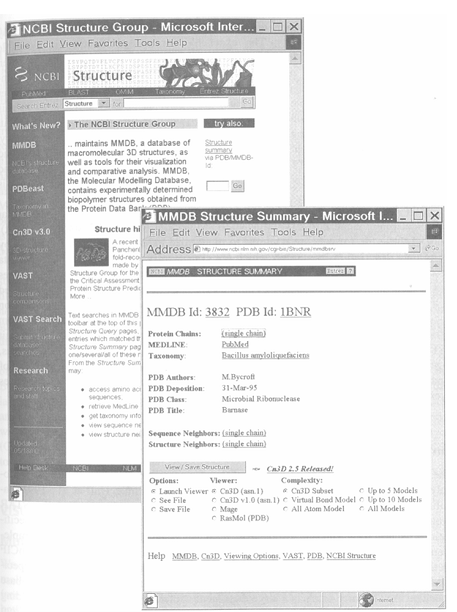
Fig.4. Structure query from NCBI with the structure 1BNR.
13.BLAST Against PDB Sequences: New Sequences Similarities
BLAST會根據使用者提供的序列 (即query) 與指定的資料庫,自動選擇適當的程式來做資料庫搜尋。是經典的雙序列比對程式,但資源消耗較大。相對的,在FASTA 中,會插入空隙以連接數個相似區(對角線),因此在最後會列出「一個」最相似的區域。Blast 並不試圖連接各相似的片段,換言之它不允許空隙的存在,所以它會計算「每一個」相似區的得分,並將此序列中得分最高的片段的分數列出。因此FASTA發現相同序列的比例低,可是它們都發生在重要的位置。
14.Entrez Neighboring: Known Sequence Similarities
BLAST的Entrez除了可搜尋sequence之外,也可藉由Entrez’neighboring facilities找出相似的DNA sequences, protein sequences,MEDLINE documents。
STRUCTURE FILE FORMATS
PDB
mmCIF
MMDB
15.Visualizing structural information
Mutiple Represention Styles
通常會利用多種圖像表示不同觀點所看到的蛋白質結構。但是依數據描繪圖像:族群、退化及動力學卻可能有盲點存在。藉由X-ray及NMR技術可同步化分子,構築出蛋白質的三維結構。這是利用晶格及核磁共振的方法所測得的。但是degenerate coordinates仍無法由資料庫的數據獲得。例如:某一原子因為相互作用的關係可能有不只一種的結構型態,對於一群分子間互相影響所形成的結構是很難預測的。典型蛋白結構如圖5。
Fig.5. A constellation of viewing alternative using RasMol with a portion of the barnase structure 1BN1.
16.Picture the Data: Populations, Degeneracy, and Dynamics
利用多維核磁共振及X-ray繞射分析法解出protein的三維結構,可求得其結構與功能之關係。X-ray 與 NMR 所預測是有結構差異的,在NMR是預測結果,而X-ray是解出結構,預測結果與實際解出之結構二者差異最大的地方是預測的結構中指出的情形,而實際的結果卻不一定存在。雖然序列特異和序列非特異蛋白的胺基酸序列相似度非常高,但由於NMR與X-ray分析的方式不同,因此有可能造成結果上的差異,加上NMR結構中並不包括DNA,因此會忽略兩分子間的交互作用。此外,局部的序列差異也有可能是造成預測結果與X-ray結構出入的原因。
17.NMR Models and Ensembles
圖6左方是X-ray形成的結構,右方是由NMR結構形成的,顯示由不同方法形成的結構會有所不同。

Fig.6. A comparision of three-dimasional structure data obtained by crystallography(left) and NMR methods(right), as seen in Cn3D.
18.Correlated Disorder
通常X-ray結構只有一個模型。然而,一些原子可能會有correlated disorder,許多X-ray結構都會將它顯示出來。但是這些3D分子圖像軟體常會忽略correlated disorder與總效果,一些軟體只秀出總體的第一個模型,或是每一原子在correlated disorder的第一個位置,而忽略掉其他。有時候更糟的是,一些錯誤的鍵結也會被畫上去,造成結構一團混亂。
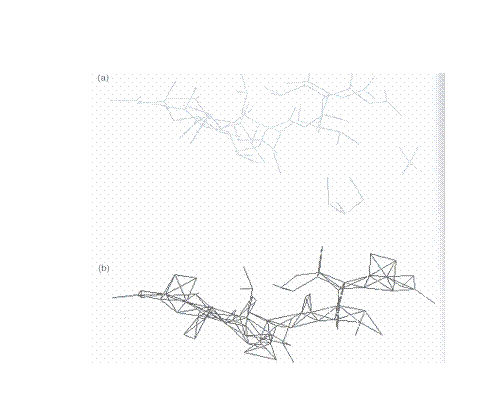
Fig.7. An example of crystallographic correlated disorder encoded in PDB files.
Local Dynamics
一般來說,內部的原子或是骨架之原子大多是由一些交互作用固定而有一致的NMR或X-ray data,而表面的原子則較有構形自由。內部的蛋白質側鏈,整體來說通常具較少彈性,因此可能的結論是蛋白質內部缺乏構形動力。然而,有一個更敏感的生物物理方法-時間決定性的單一tryptophan 殘基之螢光顯微鏡,具有可偵測到tryptophan 殘基構形的異質性(非真正的座標)。
19.Database structure viewers
在過去幾年裡,用以解釋展示結構訊息的軟體,在形象化的品質上已大大地改進,更重要的是,已經可以將序列訊息與結構訊息作相關連結。
雖然RCSB Web site 提供了RCSB data一個Java為基礎的3-D程式類型1,但是目前卻不支持非蛋白質結構的展示。因為這個與其他種種原因,常常使用RasMol v2.7取代RCSB Web site,來檢視自RCSB下載來的結構data。若較偏好Java-based viewer則使用WebMol(Figure 5.3)。
21.RasMol and RasMol-based Viewers
如上所提,現在已有一些可用來檢視PDB 檔案的觀看裝置(Sanchez-Ferrer et al., 1995)。最受歡迎的一個就是RasMol(Sayle and Milner-White, 1995)。RasMol處理PDB檔案非常謹慎,而且常常再驗算所得訊息,造成以database為基礎的不協調。它並不會試著去確認由PDB 檔案所譯成的序列或結構的化學圖形是否有效。而RasMol 2.7.1則較有改善,可以去展示整體的correlated disorder訊息,與選擇不同的NMR模型。它也可以讀mmCIF格式的3-D檔案,因此是這樣的data的觀看裝置選擇。
RasMol 含有許多極佳的輸出格式,可用以Molscript程式(Kraulis, 1991)去製成非常棒的PostScriptTM帶狀圖,用以出版。另外尚有一些程式,可免費地供學術上研究3-D結構,包括ChrimeTM,WebMol等。
Cn3D代表see in 3-D,是個用以觀看MMDB data紀錄的3-D結構觀看裝置。因為兩個原因,Cn3D一貫地具有展示3-D database 結構的能力,沒有讀取PDB檔案所需的分析、確效與處理例外的能力。(1)PDB檔案中含糊不明的data都被移去,形成MMDB data,(2)所有的鍵結訊息都很清楚。
Cn3D依靠MMDB中ASN.1紀錄的完整化學圖像訊息,因此無法讀取PDB檔案。Cn3D比其祖先具有更多特性,現在它允許選擇幾套分子的結構,與針對某特性有獨立的設定,與不同的顏色。Figure 5.1 與5.6即是來自Cn3D,根據OpenGL 3-D的圖像。
此外,Cn3D的獨特性在於它讓3-D結構變的活潑,彷彿有生命一樣。這對看NMR結構或是一連串蛋白質結構移動、蛋白質摺疊的時間變化特別有用,彷彿在欣賞一部電影或是一部漫畫。而且這項活潑的特點也使Cn3D,當配合了VAST structure-structure comparison system時,提供了較佳的多樣合作的結構。
23.Other 3D Viewers:Mage、CAD、and VRML
有一些檔案格式已經被用於呈現那些缺乏化學特異性的3D生物分子結構。他們以一般的3-D data viewer觀看結構,例如:用於macroscopic data的engineering software或是virtual-reality browsers。檔案格式,例如:VRML含有3-D圖像展示的訊息,但對於一個分子的化學基礎圖像訊息卻很少,甚至沒有。此外,這樣的檔案也很難譯出多種不同的表現風格。它需要另一個分開的VRML檔案才能呈現一個分子的space-filling model、wire-frame model、ball-and-stick model。
一些生物分子的3-D結構database 紀錄與macroscopic 軟體工具並不相容(例如以CAD software基礎的)。以電腦為目標的設計軟體呈現出一成熟的、健全的技術,一般比可獲得的分子結構軟體優秀。然而,CAD software與檔案格式一般都不適合用以檢查分子世界,因為缺乏某些專門特徵研究的觀點,與建立在檢查蛋白質細部分析的功能。
24.Making Presentation Graphics
為了自任何一分子製圖法軟體中,得到最好的出版品質圖片,首先考慮一個位映像(bitmap)或是一個vector-based 製圖影像是否需要。位映像(bitmap)是由像RasMol、Cn3D這樣的程式所製成,如螢幕上所看到的,通常其映像點就是問題來源,如Figure .7所示,其位映像為380-400 pixels。高品質的列印解析度為300-600 dots/inch,但螢幕所能顯示的通常只有72 dpi,因此,當以相同解析力的印刷機列印,一個大的影像在螢幕上會變的很小。若將影像擴大去適合頁面大小,則造成對角線的過分擴大。最好的建議就是盡可能使用大的螢幕(桌面),將映像點數(包括在影像中)最大化。
25.Advanced structure modeling
用以推敲形象化的工具,現在都已出現並可獲得。生物學家常常想要去展示出電荷分布的結構、易受影響的表面、與分子形狀;他們也想執行簡單的發展突變實驗與較複雜的結構模型。如Figure 5.8所示,SwissPDB Viewer也稱為Deep View是一個免費提供學術研究,且符合這些需求的工具。它是一個多平台(Mac、Win、Linus)的OpenGL-based工具,具有產生分子表面的能力,並可將多個蛋白質連接起來,也可快速地做出模型包括site-directed mutagenesis與更複雜的模型,例如loop重建。這些特性使SwissPDB Viewer可以做出非常美麗的圖,非常適合作為期刊封面。

Fig.8. SwissPDB Viewer 3.51 with OpenGL, showing the calmodulin structure 2CLN.
26.Structure similarity searching
雖然一個處理序列-序列相似的程式可以組合兩個序列,一個處理結構-結構相似的程式卻會造成3-D結構重疊。這種重疊導因於一套3-D rotation-tanslation matrix oreration將結構相似的部分疊在一起。一個常見的序列重疊來自於3-D重疊,藉由找到蛋白質骨架中-carbon重疊在空間中的位置。
結構相似搜尋服務是根據,兩結構間一些相似性是可以電腦計量的,並用以評定它們的相似性。這些結構相似搜尋服務可在PDB找到-比較結構、重疊database中其他3-D結構,不需知道序列即可知道最符合的結構。若兩個相似無序列相似性的結構被找到了,可知會是一項驚人的發現。為了要使這樣的數據是可以利用的,相似性的計量必須是有意義的。
目前在網際網路上可以找到一些結構相似搜尋系統而大部分這些都可在RCSB Structure Summary page找到連結。這些系統可符合各種不同需求,自人類操作的,如SCOP system,到RCSB提供的全自動的DALI、SCOP、CE system等等。
至於the Vector Alignment Search Tool(VAST;Gilrat et al., 1996)則是提供了3-D結構的相似性測量。它使用來自二級結構的vector,搜尋時不需要序列訊息,因此當沒有序列訊息可偵測時,VAST可以找到結構相似性,如同SLAST以N×N方式搜尋database,其結果可以儲存並使用Entrez 介面快速取回。目前,以VAST演算法可以比較database中超過20,000 domain structures,得到結構-結構重疊的紀錄,並組合來自重疊結構的序列。VAST演算法焦點在在統計上令人驚訝的相似性,不浪費時間在檢查許多小的次結構相似性,因為這在結構比較中發現只是偶然出現。舉例來說,-sheets常常出現,但這並不是什麼令人吃驚的相似。因此,他們提供的是較廣的觀點,針對protein family的結構、功能、與演化。
VAST在這麼多比較的工具中表現突出是因為:(1)它有清楚定義的相似性計量,可以找到令人驚訝的關係,(2)具有可調整的介面,可在最有興趣的關係中快速觀看,不需要看很多遍相同的關係,(3)它提供了一個domain-based 結構比對,而非整個蛋白質比較,(4)可與Cn3D合併作為形象化工具,更詳細地檢閱令人驚訝的結構關係(Figure 5.9)。
除了列出相似的結構,VAST-derived structure neighbors含有細部的residue-by-residue組合,與3-D 轉換矩陣變成結構的重疊。藉由VAST superposition,我們可以鑑定哪一區域在蛋白質演化中被修飾過,而DALI(Holm and Sander, 1996)superposition對於牽涉到marking structural model的比較會較有用。不管如何,VAST與DALI superposition都是研究蛋白質結構關係極佳的工具,特別是與SCOP protein family database一起使用時。

Fig.9. VAST structure neighbors of barnase.
27.參考資料
1.
http://binfo.ym.edu.tw/post/
2.
http://www.37c.com.cn/topic/004/netguide/netguide01.asp?filename=cn3d.htm
3.
http://www.cbi.pku.edu.cn/chinese/documents/softdoc/stru.html
4.http://strbio.biochem.nchu.edu.tw/classes/SF%20student's%20web/2001-8%20TakeHome/HMG-D/right-e.htm
5.
http://xcc.life.nthu.edu.tw/Keywords.htm
28.Internet resources for topic presented
BIND http://bioinfo.mshri.on.ca
Imagemagick http://www.wizards.dupont.com/cristy/ImageMagick.html
mmCIF Project http://ndbserver.rutgers.edu/NDB/mmcif/index.html
National center http://www.ncbi.nlm.nih.gov/
for Biotechnology Information(NCBI)
NCBI Toolkit http:// www.ncbi.nlm.nih.gov/Toolbox
Nucleic Acids http://ndbserver.rutger.edu/
Database(NDB)
POV-RayAY http://www.povray.org/
Protein Data Bank http://www.rcsb.org/
At RCSB
RasMol http://www.berstein-plus-sons.com/
SwissPDB Viewer/ http://www.expasy.ch/spdbv/mainpage.html
Deep view
WebMol http://www.cmpharm.ucsf.edu/~walther/webmol/