應用核磁共振研究蛋白質的構造及動態
口述:黃紹光 博士
整理:黃信明
今天非常榮幸有這個機會到貴校來,來和各位一起研討蛋白質體學這門重要的課程。大家都知道,現在是新世紀剛開始的幾年,過去的那個世紀一般科學家都把他叫做物理學的世紀,因為在二十世紀初一些物理理論相繼出爐,到了後期大家逐漸對生命科學的研究產生興趣。當然生命科學本身是門很奧妙的學問,以前也許是因為各方面的理論或適宜器方面都不是這麼精確,所以研究生命科學有很多困難,但是隨著物理學以及其他學科的進展,大家又開始回到生命科學的研究,所以到了現在對於生命科學就以不同新的方式研究,所以大家就開始講現在為生物的世紀,這在全世界很多地方都可以看得出來。
今天我們聚集在一起,也就是一起研究生命科學裡面很重要的一環,也就是蛋白質體學,這個方面需要有許多不同領域的人來共同研究,所以很多國家與大學有花很多資源在上面,如中研院有成立一個center of genome research;而貴校成立一個生命科學系的意義非常重大。
今天我界這個機會和大家探討研究蛋白質體學的方法之一,主要就是利用光譜學來研究蛋白質的構造,所以我的這個題目就是以光譜學來研究蛋白質結構及動態。蛋白質之所以重要,在生物體中蛋白質是組成所有細胞、組織的基本物質,那這個生物體如何合成蛋白質,這完全看gene,通常由三個鹼基決定一個胺基酸,所以有64個組成,genetic codes可決定蛋白質是由哪幾種胺基酸所合成的,當然長度也是由genetic codes決定,最後由terminatus結束。蛋白直數量相當多,在生物體中應該超過百萬,每一個蛋白質的大小與功能都不一樣。
有些比較簡單的生物體的蛋白質量是比較複雜的,這個表示顯示不同的生物體的蛋白質大小是用幾個胺基酸來表示,有的非常大的蛋白質有大於六千個胺基酸,有的則比較小一點。NMR本身有許多先天性的不足,跟X-ray相比有許多比不上的地方,這個曲線代表的是說到目前為止NMR能夠研究的一些蛋白質都是比較小的,太大的無法研究,因為太複雜了。
剛才講過全世界每個地方對於gene、蛋白質的structure都非常的注重,像美國NIH就支持九個很大的研究中心研究protein structure,有些是專門用X-ray研究, 有些用NMR,而有些是兩個都有的,不只是美國,日本也是一樣有在研究protein structure。
在國際上有一個Data Bank,在世上有做研究的都會把結果送到這個Data bank裡面,各位可以看到從這個表格以經解出的蛋白直數量隨著時間增加,可以想像之後幾年解出蛋白質的數量將會非常的多,蛋白質可能有一百萬或是更多,所以要完全瞭解各種不同蛋白質的功能需要的時間還是很長,那麼每一個實驗室有很多的儀器,能夠解出蛋白質的結構數量非常的少。
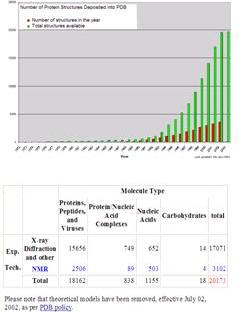
這個表格列出來的就是利用兩種不同的方法解出蛋白質,一個是X-ray diffraction,另一各式NMR,目前為止此二法為有效的方法,X-ray能解出較多的結構,而NMR所解出的蛋白質結構佔不到五分之一。
我們研究蛋白質的構造,通常描述蛋白質的結構有幾個層次,第一個層次就是看sequence,看是由哪幾個胺基酸構成,順序是如何,NMR最主要研究第二及第三層次的構造,即研究蛋白質的骨架,他的形狀主要是因為有氫鍵、carbon、oxygen,把這個backbond變成特定的形狀;第三層包括不只骨架,涉及到side chain的形狀如何,這對蛋白質的功能也很重要,很多side chain為聚水性;再下一層是只有很多很大的protein有很多的subunit互相之間的關係,通常這方面NMR比較難研究,因為整個分子量太大。
除了這種結構以外,蛋白質的功能跟她的動態關係非常密切,這動態包括conformational freedom,所有的結構有哪些地方比較固定或鬆散,骨架有時不是很固定還有proline cis-trans ismerization,這些conformational freedom是其中之一;第二個是folding,protein如何造成固定的形狀,如何摺疊起來變成特定形狀,這過程可以用NMR來研究;第三個就是binding,protein和大分子或小分子如何結合、結合速度、結合出的結構為何?這對蛋白質的功能也非常重要。
剛剛有提到為何蛋白質的動態,因為蛋白質的動態(protein dynamics)和蛋白質的功能很有關係,所以蛋白質在發生和作用時都會牽扯到他的motion或整個結構的改變或和小分子如何結合,另外很很可能尤其在水中蛋白質的結構可能有很多其他的未知結構,然後互相的換來換去,其中也許只有一種結構跟其功能有關,所以研究蛋白質結構時得注意到這點。
剛剛講到光譜學來研究蛋白質的研究,事實上光譜學是跨領域的學問,今天主要講到NMR的方法。我們剛看到的這個表格知道幾乎所有解出來的蛋白質結構都是用X-ray或NMR來做,所以大家可以比較NMR和X-ray兩種方法有何不同?首先,NMR解出的結構通常可以在solution裡面,這比較接近protien在細胞裡或生命體內的環境,而X-ray通常是解固體的結構,當然一般來講大部分的蛋白質的固體結構和溶液中的結構相當類似,但有可能會有部分的地方不太一樣。另外NMR可以用來研究他的動態,X-ray要研究動態比較困難點。第三點就是NMR的靈敏度不夠,解析度也有限制,所以現在幾乎大的protein都無法用NMR來多,一般來說是五萬個Dalton以下可以用NMR來解,以上的話就得看情形,但X-ray的限制比較小。
到底NMR是做些何事?說穿了就只有三個參數:chemical shifts、coupling constants和relaxation times,也就是不同的原子核在光譜出現的位置不一樣,為何不一樣?可能是因為原子核本身就不一樣,當磁距不一樣,其在磁場中旋轉就不一樣,再細看因化學鍵的不同會產生不同的電子雲,也會影響其在磁場中的顫動,另外原子核的鄰居也會造成原子核周圍電子雲散佈的不同。
NMR的基本觀念通常用這圖形表示,one half的意思就是他會有兩個能階,這兩個能階的能量若不放在磁鐵裡的話是一樣的,妳放在磁鐵裡後能階就會隨著磁場的大小散開,所以在一定磁場其位能是一定的,位能和磁場強度和自己磁距的大小有關係,做NMR實驗就是想辦法供給下面的原子核一些能量讓他跳到上面,使他吸收能量後可以再光譜出現。
在一磁場中1H為900 MHz,而13C因磁距比較小所以為226 MHz,15N為更小。細看同樣是proton因為化學鍵不一樣或周圍環境不一樣,分散在13500 Hz的範圍之內,13C的範圍就更寬了,有200 ppm通常,要看是哪一種碳而分佈在不同的地方;15N也是一樣,他的範圍為30ppm,是因為他的化學鍵的不同或是附近環境不一樣而造成。做NMR實驗通常事看氫的光譜,因為他的氫力高,他的靈敏度也高,這是很典型的一個蛋白質的氫光譜,可看到很多的訊號在這裡,可以想像如果妳有一百個胺基酸,會有多少個氫原子在裡面,每一個都有獨特的訊號,加起來就一團糟,我們大致上可看到在這個範圍之內的為beta proton,在3.5到5大概為alpha proton,而芳香族的proton則大致在6到10,有的甚至到更低的地方,所以大體從這個出現的位置我們可以知道是哪一種proton。
最近就是因為解出的蛋白質比較多,所以從解出的蛋白質proton訊號統計的結果可以看出來這些胺基酸的alpha proton等的大致位置。另外同時也從解出的蛋白質內看到,不同結構-HN出現的地方不太一樣,這是一個很好用的地方,假如說妳有一個不知道是何結構的蛋白質,妳看出其-HN出現的地方是在哪個地方,就可對照這個表可預測他大致的形狀及結構為何,下面幾張投影片的alpha proton、N都有類似的趨勢,所以知道這個結構的protein越來越多,我們得到的訊息也就越來越多,所以現在可以寫出一個程式來預測一個protein的結構為何,可以從chemical shift來預測,但就目前來說準確度尚可。
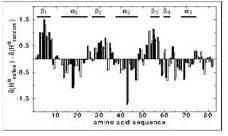
下圖是個例子,這個protein叫Glutaredoxin 3,他有八十幾個胺基酸,看其alpha proton的chemical shift和random coil的平均值比較,從第一個位置到第十個可以看到alpha proton都比平均值高一點,所有從比較高的部分從剛才的表格可以預測為beta sheet的形狀,比較低的可能是alpha helix。
那第二個參數為coupling constants,即兩個原子核之間的交互作用,這個交互作用主要是由dipolar,就是磁距和磁距之間的交互作用,這個是看兩個原子核距離多少而定,非常的靈敏,和距離的六次方成反比;若樣品為固態,因其作用有方向性,所以造成光譜非常的寬,但在溶劑中,這個方向性就平均掉了,做NOE實驗可以決定兩個氫原子中間的距離大致為多少,所有解蛋白質結構都得靠這個實驗;第二個交互作用為indirect J-couplings,就是原子核經過chemical bond上的電子,而間接影響到鄰居的原子核互相作用的大小,這個coupling通常在氫光譜中很容易發現,因為看旁邊有幾個不同的proton,本身的proton的形狀會改變,此外coupling大小和conformation也有關係。
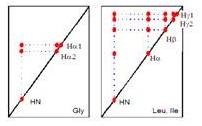
接下來看到二十個胺基酸因為氫原子含量不一樣而光譜的形狀不一樣,glycine只有兩個alpha proton,因兩個proton經由J-coupling造成AX,Ala因為是CH3所以為A3X;每一個胺基酸有特定的pattern,這各式很重要,因妳得到氫的光譜妳得知道哪一個是哪一個proton。
這個隔著三個chemical bond的carbon,這個大的圈圈是nitrogen,和nitrogen接的為NH,穿過nitrogen看這個alpha carbon虛線的圈圈接的是alpha proton,所以妳把nitrogen的alpha carbon從前面到後面排列的時候可看到兩個proton之間形成一個角,coupling constants就是這兩個coupling和角度的關係,假如角度是0為8 Hz,90度幾乎是0,所以從量出了J-coupling你就可以決定兩個proton之間的夾角為多少。
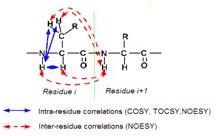
這也是圖表兩個胺基酸,我們做NMR需要得到的事correlations,主要是有兩種關係,一個是J-coupling另一個為dipolar coupling,同一個胺基酸妳會發現他的correlation為一個NH和一個alpha H,這個藍色部分就是indirect correlation,所以這互相之間可能是J-coupling的關係也有可能是dipolar的關係。我們可以做Correlated spectroscopy (COSY)的實驗來看它們之間到底有無J-coupling,bipolar coupling可用Nuclear overhauser effect spectroscopy (NOESY)來做,所以做NMR實驗主要是COSY和NOESY。
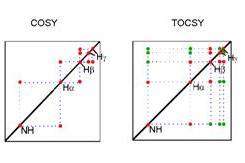
下面是一個例子,像一個胺基酸用COSY,NH、alpha、beta、gama分別位於這裡,因為NH和alpha有couple,所以做了COSY實驗可發現相對於這兩個會有對角現之外,表示說NH和H-alpha有J-couple,同樣的alpha和beta也是;Total correlated spectroscopy (TOCSY)是另外一種,這個很重要因他可告訴你哪些是屬於同一個胺基酸,只要是同一家的proton都可以看到這個訊號,所以NH和H-alpha、H-beta都有一個訊號,當然這會造成圖形很複雜,當經過這個分辨就可以分辨其是否屬於同一個胺基酸。下面這一張的每一點代表一個胺基酸,比如說這是某一個胺基酸的proton signal在9.3的地方,所以只要算幾個點就知道有幾個胺基酸。
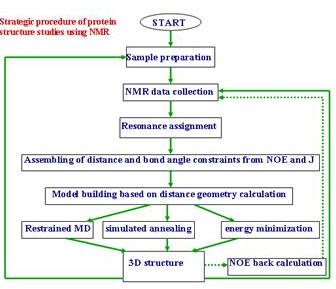
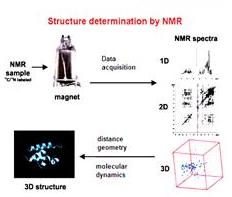
這個流程圖就是用來指導你做NMR實驗怎麼做,為了要決定這個蛋白質的結構,第一個步驟就是要把樣品做好,最好是把他純化,有一點雜質是無妨,因剛看到TOCSY可以把不同的胺基酸分出來。妳得到sample以後就把他溶在溶劑裡就開始做NMR實驗,先做簡單的NMR再做COSY、NOESY,下一步是要決定哪一個peak是屬於哪一個proton,妳看出來之後就可以知道proton和proton之間有沒有NOE;下一步就是把結果送進計算機決定構造。
樣品的準備,首先是純化,之後選正確的溶劑,溫度和酸鹼度得調好,假如分子太大或是有其他困難地方,得合成類似的物質,最後就是isotope labeling。
Assignments怎麼做呢?剛剛提到1D proton NMR、2D COSY and TOCSY;另外還有很多實驗可做,NH exchange rate,這可以看出哪一個amino acid的NH是可以很容易exchange;temperature dependence和氫鍵有關係。
這個圖表就是告訴我們怎樣利用NOE來決定哪一個proton屬於哪一個amino acid,當一個proton和另一個proton有NOE,表示這兩個是接近的,通常鄰居的amino acid的NH距離很接近而產生NOE的現象。COESY看到的是同一個胺基酸內,當妳知道一個proton在哪裡,你就可以知道下一個alpha proton在哪裡,妳做一個NOE後,妳這個amino acid的alpha proton和鄰居的NH proton會有NOE,所以你就可以從這個proton決定這一個,依此類推。
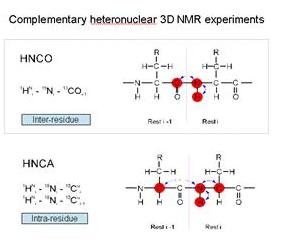
有些實驗可以造成不只像二維的NOESY、COSY的NMR圖形,還有很多可以做三維的實驗,HNCO這叫inter-residue correlations;類似的HNCA叫intra-residue,看這個胺基酸本身,妳可以做一個3D的圖譜。下面這張就是很典型的HNCA NMR的圖譜,每一個是代表這個胺基酸intra-residue。下面這個HNCA,intra-residue correlation妳可以找到proton的位置,然後妳做一個HNCO妳可以找到他前面一個coupling-carbon的位置,依此類推。
事實上做實驗時,妳得到一個3D的光譜,妳必須像切豆腐一樣在有訊號的地方切下來,在那個地方切下來會有一個訊號,這個訊號會告訴你那個amino acid位置的CO為多少,所以妳切的很多然後安排妥當後,你就可以決定他的proton signal是在哪裡,這個步驟非常的繁複,通常一個學生要花好幾個禮拜或好幾個月把這個圖譜做好。
下個步驟,妳知道哪個訊號是屬於哪一個proton以後,就要決定結構了。NOE有時候太複雜,所以妳得想辦法將NOE的圖譜簡化,簡化可能牽涉到比較高維的NMR實驗,最後把這些NOE的大小決定後,妳可以送到computer內做一個model,另外在用很多其他牽扯到molecular dynamics或者是其他方式將結構定的更細,另外一點就是你定完結構以後,從定好的結構還可以反過來換算NOE是多少。決定結構最主要看NOE,因為NOE和距離有關係,胺基酸本身在排列時會有不同的構造。
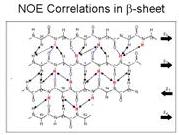
所以做好實驗後,把所有的NOE排列起來,當proton鄰近的NOE很強的,大致上為beta-sheet結構,假如是NH和NH NOE很強,NH和alpha隔了三、四個amino acid的話,代表說這個部分是一個helix。Beta-sheep的coupling constants很大,有時候會九點幾;alpha-helix就比較小。假如一開始你不知道哪個proton和哪個proton的NOE時,你就無法決定他的構造。經過distance geometry後,可以得到一個structure,最穩定的結構應該是能量最低。分子大的時候需要的資訊越多,有一些實驗得到的參數是和氫鍵的長短有關,氫鍵很短表示CFA比較大,可以預測構造為beta-sheet。
剛才我們都在講結構如何決定,那動態如何研究呢?首先我們看到底動態包含何東西,第一個就是binding,和小分子binding,通常我們做實驗的時候可以把ligand從沒有慢慢的加入,看結構有何不同,另外可以看NOE可以找到binding site,第三個實驗室transfer NOE,有時候小分子的binding不是很強,這個實驗可以決定protein哪一個地方和ligand連接;接下來NH exchange rate,NH proton本身會和其他的proton交換,或者和水交換,這和dynamics也有關係,另外一個就是鬆弛的時間,T1是比較快的,下面還有一個就是解釋結果,可用modelfree來解釋,假如這個參數是1表示穩定,0是表示結構不穩定。Dynamics的速度不同,所以用的NMR實驗以不同,很快的時候用order parameters可以大致決定哪一部份的蛋白質結構比較穩定,不穩定的通常牽扯到side chain,稍微慢一點可以用relaxation time來決定,更慢的可以看T1ρ,在更慢的話妳會發現他信號本身會改變,有的比較寬有的比較窄,有exchange的話信號會比較寬的現象,非常慢的話妳會發現有兩種不同的構造出現在光譜上。
分子越大的問題越多,很多實驗室都在想很多不同的實驗來解決這些困難,最基本的就是分子大的光譜會複雜,用更高的磁場解析度會比較高,圖譜會比較簡化,可以用isotope filtering將信號簡化;假如分子太大,信號鬆弛的太快,想辦法鬆弛的過程有許多是互相抵觸的,利用抵觸的部分把鬆弛的時間增長,所以每個峰都比較窄,得到的光譜比一般的光譜還要尖銳,他事實上因這個發明得到去年的諾貝爾獎。CD (Circular Dichroism)主要是看不同的左旋、右旋,不同的光打在裡面的吸收度不同而決定他是含哪一種結構。
我講的題目本身跨了很多的領域,當然生命科學本身要研究的方法很多,所以方法很多,之後的演講者講的會更精彩,謝謝各位。