蛋白質體學概論
口述:陳義雄 老師
整理:黃國展、傅綜信、劉政嘉、張春揚
任何時空文明的進展都是由事實的收集成為知識,再累積成為智慧。將智慧表達出來,即是各個時期的文明;而自從分子、原子的發現之後,科學的進展也進入了另一個時期。此外各個學科的表達方式也有不同,像文學家、藝術家可以在作品中表達個人的情感,而一個科學家或許會因情感而有不同的作法,但不能因此而影響他研究的客觀性。科學研究是要經得起邏輯分析的。
在20世紀中,分子層次探討的生命科學和相對論、量子力學合稱本世紀最主要的三大科學文明。這些都是從基礎的研究再慢慢的進入應用的階段,而其中從分子層次開啟研究的生命科學所發展出的已成為跨領域的應用,包括電子業、機械業。目前生命科學的研究已進入後基因體時代,也就是說基因序列都已經知道了。若先不考慮相關的調控,我們可以把重點分為兩類:一、functional genomics,也就是研究這個基因俱有怎樣的功能,若是從蛋白質的層次來看的話,我們便要有蛋白質體的研究。二、structural genomics,則是研究基因產物的構造。那為什麼要有蛋白質體的研究呢,這是因為蛋白質和基因之間並不是單純的線性關係,基因只提供要產生何種蛋白質的基本序列,就像食譜一樣,但是我們利用不同的烹調方法來決定我們的食物,因此無法只從基因的序列就瞭解蛋白質所有的特性。
若把一個細胞當成一間房子,那麼房子的磚頭、水泥便是蛋白質。蛋白質雖然是經由基因表現而來,但對細胞而言,蛋白質才是真正執行功能的物質。因此目前的研究便是集中在所謂的human proteome,人體中由細胞或組織合成的所有蛋白質。
1、研究基因的功能目前主要有兩種方法:
(1)gene knock in/out,也就是在個體中放入某個基因或破壞某個基因,再看個體的反應,然後觀察該基因的功能。但是有些反應外觀看不出來,需要到分子層次才看得出,表示這方法也有其盲點。
(2)proteomic,以生物化學的眼光大規模地研究某一蛋白質的特性,如在細胞中的活性和其參與的生化途徑。
2、蛋白質體研究和基因體研究的差別:
(1)蛋白質體的研究比起基因體更為複雜,因為人類的基因體大約有4萬個基因,但一個基因有時可以表現出多種蛋白質。
(2)直到目前為此,還有30%~50%的人體蛋白質不知道功能,且蛋白質需折疊成特殊的立體結構才有功能。
(3)而基因表現出來後還有所謂的post-translational modification,因此同一蛋白質在不同的環境下會有不同功用。
主要是研究基因上的open reading frame是否有表現出便產生蛋白質。藉由大規模的分析,鑑定蛋白質的微量特性。以及該蛋白質因受到post-translational modification,造成不同的同份異構物,並加以分析比較兩者間的差異。從正常與不正常的細胞之中分離出相關的蛋白質體,再來互相比較,可看出疾病會對此蛋白質體之中的哪些蛋白質有影響,而此蛋白質即可作為該疾病研究的特徵。因為通常功能相同的蛋白質會聚集在一起,因此可知道蛋白質如何在一細胞內構成一網絡架構,最後可研究蛋白質的立體結構,來瞭解它如何結合藥物及藥物如何增強或減弱這蛋白質的活性,這樣將可減少藥物的副作用,並可研究蛋白質在訊息傳遞或代謝途徑中活性的改變。
1、 2-dimensional electrophoresis
(1)2-dimensional electrophoresis的原理及特性:
(a)利用第1 圍為蛋白質的帶電量(electric charges)及第2 圍為分子大小(molecular sizes)不同來分離蛋白質。
(b)2-dimensional electrophoresis之前的準備工作pre-fractionation可以使分析的效果大大提高。
(c)2-D作出之Gel,我們可以利用以下幾種染色方法來做偵測蛋白質:Coomassie blue,Silver nitrate或fluorescence dye 。
三種偵測方法的比較:
|
|
優點 |
缺點 |
|
Silver nitrate |
敏感度好 |
專一性差、非線性關係 |
|
fluorescence dye |
敏感度好、線性關係 |
Degradation快 |
|
Coomassie blue |
線性關係 |
敏感度差 |
※線性關係是指螢光或顏色量與測得的蛋白質量成正比
補充:
pre-fractionation:依照分子的物理化學特性(例如:solubility、net charge, mobility, size, hydrophobicity, and affinity)來分類要分析的蛋白質。Sub-cellular fractionation 可用以分出功能相關的蛋白質。
(2)2-dimensional electrophoresis之用途:
(a)分析不含mRNA的sample。例如體液。
(b)蛋白質表現量與其mRNA量不符合。(即使有很多mRNA,不一定會表現出與預測該表現的蛋白質量一樣。)
(c)可偵測出蛋白質經Post-translational modification(例如:Phospholation、Glycosylation、Hydroxylation…等)活性的變化,而不只是蛋白質量的變化。
(d)對於一個重要的製造蛋白質來源作一分析。
(e)研究微生物的proteome。
(f)比較兩個不同的proteome,利用顯示出來的2-D gel 上的相異處。
(3)2-dimensional electrophoresis的限制:(1)太大或厭水性的蛋白質無法用此作分析(2)無法分出全部的蛋白質(3)無法定義正常蛋白質的表現為何。
2、Mass Spectrometry:
根據每個蛋白質的荷質比不同,使用電場與磁場來分析蛋白質-Mass spectrometry。Mass spectrometry主要由三個系統組成。分別為ionization source,mass analyzer,detector。
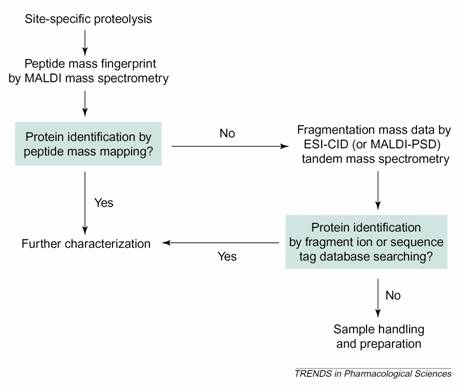
圖1:蛋白質經過酵素處理作用,利用MALDI mass spectrometry 加上已知的蛋白質database比對並找尋未知的蛋白質,如果第一次不能成功從database 找到我們要的蛋白質,可以利用tandem mass spectrometry再配合database找尋相關的蛋白質,如果仍然不能將未知的蛋白質的身分鑑定,可以考慮重新純化或處理未知的蛋白質,再從頭開始(1)。
整個過程可以由圖1來說明,蛋白質先經過site specific proteolysis(利用酵素俱專一性切特定的序列),進入MALDI(matrix-assisted laser desorption/ionization )mass spectrometry,利用實驗得到的質譜,從database找尋相似的質譜,即可定出此段蛋白質的序列,就完成mass的分析。如果分出的其中一fragment仍不能配合database找到相似的蛋白質質譜,就要再做另一次 tandem mass spectrometry(ESI-CID或MALDI-PSD), 再進一步去分析,如果可配合database而定出序列,便完成mass的分析。若還有不能定出之fragment,反覆做mass,配合database,直到找到有可能相關的蛋白質質譜,定出蛋白質的序列。然而,利用mass spectrometry-based method進行蛋白質分析時,需透過資料庫中的資料與分析出來的數據作比較,藉此來辨識蛋白質,但有時無法尋得符合的資料,其原因可能牽涉到蛋白質的Post-translational modification。
補充:
蛋白質的Post-translational modification:
(1)Phosphorylation:蛋白質經過phosphorylation後,其mass將增加80Da。
(2)Methylation:蛋白質經過methylation,其mass則是增加14~15Da。
(3)Lipidization:蛋白質加上lipid。
目前mass spectrometry-based method中較易解決的是phosphorylation的問題,其中threonine、tyrosine及serine的phosphorylation已有specific antibody可用來鑑別。若懷疑蛋白質是否有phosphorylation,可利用mass spectrometry-based method去分析有經過phosphotase處理與未經處理的蛋白質,經過phosphotas處理過的蛋白質其mass會減少80Da,因此可由圖譜中發現有向左位移的現象,如圖2所示:
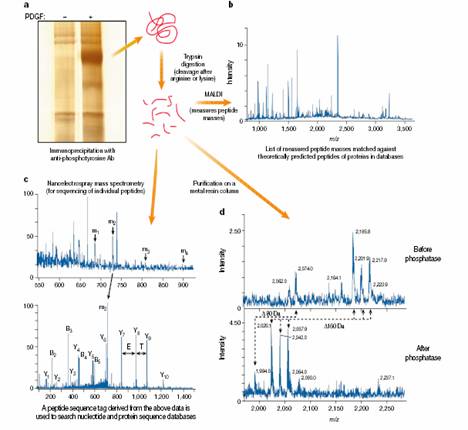
圖2:圖a. 將細胞經過PDGF處理後,再將cell lysates利用anti-phosphotyrosine antibodies進行immunoprecipitation。經過one-dimensional gel electrophoresis,將gel進行silver stained,之後再以trypsin進行digestion。圖b. 經過trypsin處理後的蛋白質利用MALDI進行蛋白質分析比對。圖c. 經過trypsin處理後的蛋白質,亦可利用nanoelectrospray tandem mass spectrometry進行individual peptides的序列分析。圖d. 經過trypsin處理後的蛋白質,還可利用metal resin micro column進行蛋白質純化,之後可再經過MALDI或nanoelectrospray分析未經過或已經過alkaline phosphatase處理的蛋白質,並可發現經過phosphotas處理過的蛋白質其mass會減少80Da,因此可由圖譜中發現有向左位移的現象(2)。
3、Capillary electrophoresis:
其原理是利用蛋白質等電點的不同來進行分離,之後再藉由Fourier-transformed ion cyclotron resonance (FTICR) mass spectrometer來進行蛋白質辨識。此外,亦可利用2-D liquid chromatography(即經過兩次HPLC分析)並結合mass spectrometry-based method來進行分析,但因此方式需耗用大量的電腦程式進行運算,故研究進展較為緩慢。
以下為Capillary electrophoresis的優劣比較:
|
|
優點 |
缺點 |
|
1 |
不需由gel中取出蛋白質,可直接從溶液中取得蛋白質。 |
蛋白質須先進行pre-fractionation才能使用。 |
|
2 |
僅需作one-dimensional electrophoresis便可直接經Mass spectrometry-based method進行分析。 |
|
4、 X-ray crystallography:
蛋白質的三度立體結構可利用X-ray crystallography的方式鑑定出來,其原理是利用一X-ray 光束打至蛋白質晶體上,經由繞射原理去推斷出結構狀態。
5、 Microcaloriutoie:
利用兩物質間的作用會產生熱力學上的變化,進而去監測兩者之間的熱力學變化,來判定兩者之間是否有interaction,故此檢測方式十分靈敏。
1、蛋白質的網路架構(Protein network):
Protein network意即蛋白質與蛋白質之間互相結合聚集,因而產生一連串的生物化學反應或表現細胞各種活性。因此未知蛋白質可利用其與已知蛋白質之間的interaction來推測此未知蛋白可能的功能。
2、蛋白質的純化方法:
(1)affinity-based method:利用glutathione S-transferase (GST)-fused polypeptides來進行分離,其原理是利用一個含有glutathione S-transferase基因的載體(vector),將蛋白質的基因接在glutathione基因之後,因此可製作出glutathione S-transferase (GST)-fused protein,當其他的蛋白質若和我們接在glutathione後面基因所產生的蛋白質有相互作用時,便會結合在一起,再來利用蛋白質上有一個thrombin 可以切的地方,可將我們想要的蛋白質從glutathione分離出來,最後經過電泳,即可分離出有相互作用的蛋白質。圖三為一操作例子,首先將glutathione S-transferase (GST)-fused protein與column相結合,圖中的Tag可視為glutathione,而S14則為glutathione S-transferase (GST)-fused protein,其可與欲分離之蛋白質相結合,故可將cell lysate通過此column,使得欲分離之蛋白質以affinity的方式與S14相結合,之後再加入protease(即thrombin),因此便可分離出欲得的蛋白質,之後再以mass spectrometry進行分析。此外,亦可利用antibodies,、peptides、DNA或RNA結合在column來進行蛋白質純化或分離。
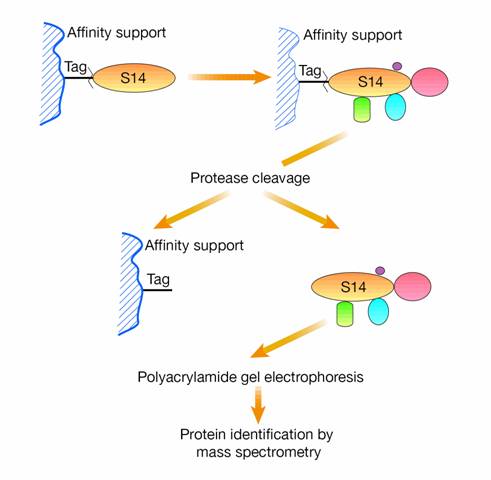
圖3:首先將glutathione S-transferase (GST)-fused protein與column相結合,而S14可與欲分離之蛋白質相結合,故可將cell lysate通過此column,使得欲分離之蛋白質以affinity的方式與S14相結合,之後再加入protease(即thrombin),因此便可利用競爭方式分離出欲得的蛋白質,之後再以mass spectrometry進行分析(2)。
(2)在yeast中有一較為成功的例子,即利用一蛋白質與一epitope相結合,並使其在細胞中過度表現,之後將細胞打破,再利用可辨識此類epitope的抗體(antibody),進行蛋白質免疫沉澱(proteins-immunoprecipitation),因而將蛋白質分離出來。
(3)一蛋白質與其他蛋白質為Low-affinity interaction,因為在分離時容易從有相互作用的蛋白質脫離,不容易被分離出來,因此以化學方法將蛋白質cross linking在其他protein complex上,如此才能進行下一步的分離純化。舉例來說,可利用photo-affinity labeling的方法將蛋白質分離,其原理是將蛋白質接上一個photo-affinity label,再加入ligand(另一蛋白質),經照光使得兩者產生covalent cross linkage,形成protein complex,進而可以進行分離純化。
(4)Yeast two-hybrid system:利用轉錄音子(transcription factor)來證明protein – protein是否有交互作用的一種技術。由於transcription factor(如GAL4)上常含有DNA-binding domain及Transcriptional activation domain,當此兩個domain 結合在一起,且DNA-binding domain結合到報導基因(reporter gene)上游的activating sequence,Transcriptional activation domain才會啟動下游的 reporter gene 表現;若在DNA-binding domain接上一個蛋白質X,在Transcriptional activation domain接上另一個蛋白質Y,當X及Y有interaction時,此DNA-binding domain-X-Y-Transcriptional activation domain complex會一起結合到reporter gene上游的activating sequence,而啟動下游的reporter gene表現,相反的,若沒有interaction,DNA-binding domain-X及Y-Transcriptional activation domain便不會形成complex,即使DNA-binding domain-X 結合到reporter上游的activating sequence,也不會啟動下游的reporter gene表現,單獨的Y-Transcriptional activation domain也不能直接啟動reporter gene,因此利用此特性,我們可以知道蛋白質X及蛋白質Y之間是否有交互作用,將GAL4的DNA-binding domain(GAL4-BD)和GAL4的activation domain(GAL4-AD)分別接上不同蛋白質後,GAL4-BD會和ORF上的GAL4-binding site相接,但因蛋白質X及蛋白質Y 之間並無交互作用,因此GAL4-AD並不會和GAL4-BD相結合,所以並無法活化reporter gene的transcription(圖4a);當GAL4-BD與GAL4-AD同時存在並互相結合時,才能活化reporter gene的transcription(圖4b),因此我們可利用兩種yeast cDNA library,分別為可產生DNA-binding domain蛋白質的ORF-BD library與可產生Transcriptional activation domain的ORF-AD library,在將此兩種菌株進行交配,之後利用deficient medium進行選殖,唯有菌株含有GAL4-BD與GAL4-AD同時存在才能存活(圖4c),如指含有其中任何一樣皆不能在deficient medium下生存,因此可從deficient medium獲得許多interacting proteins,但如此眾多的蛋白質中何種才是所欲求的蛋白質呢?這便需要一個個去進行分析才能獲得解答。
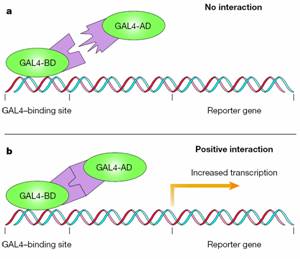
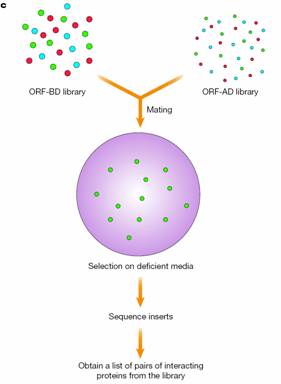
圖4:圖a. 若在DNA-binding domain接上一個蛋白質X,在Transcriptional activation domain接上另一個蛋白質Y,當X及Y沒交互作用,DNA-binding domain-X及Y-Transcriptional activation domain便不會形成complex,即使DNA-binding domain-X 結合到reporter上游的activating sequence,也不會啟動下游的reporter gene表現。圖b. 當X及Y有交互作用時,此DNA-binding domain-X-Y-Transcriptional activation domain complex會一起結合到reporter gene上游的activating sequence,而啟動下游的reporter gene表現。圖c.製作兩種yeast cDNA library,分別為ORF-BD library與ORF-AD library,在將此兩種菌株進行mating,之後利用deficient medium進行選殖,唯有GAL4-BD與GAL4-AD相結合的菌株才能存活,因此便可獲得許多interacting proteins(2)。
(4)Phage display則是利用Bacteriophage particle的方式,將有興趣的DNA的蛋白質或peptide全都產生在capsid或coat protein,之後再利用和此蛋白質有作用的peptide epitopes、peptide ligands、enzyme substrates 或antibody fragments來進行screening,捉有相互作用的蛋白質,進而分離出欲求之蛋白質。此方式與Yeast two-hybrid system的原理很相近,兩者差異在於Yeast two-hybrid system必須在nuclear中才能使用,Phage display則無此限制。
(5)蛋白質晶片方法(Protein chip approach):近年來蛋白質晶片發展十分迅速,其基本操作程序為large-scale format functional assays能夠被表現及純化Recombinant proteins,例如分析酵素的活性,而此方法亦可被稱作biochemical genomics。Protein chip則可利用不同方式製備,其晶片表面可固定一層recombinant proteins或recombinant proteins domains (例如GST–fusion proteins),之後再將cell lysates或phage cDNA display library加入晶片中,使其與recombinant proteins或recombinant proteins domains進行interaction,接著沖洗掉無法結合在晶片上的蛋白質,最後將結合在晶片上的蛋白質取出並利用mass spectrometry進行分析,或是利用放大方式將和蛋白質晶片上蛋白質有交互作用的phage particles分離出來,再進行cDNA序列分析。而yeast two-hybrid system亦可以進行蛋白質分析,首先是將yeast cells轉植入individual ORF–activation domain fusions,在將這些cells培養於培養皿(plates)或濾紙(filters)中,因此每個單位擁有一個yeast clone,其含有一個unique ORF,接著再與含有single ORF–DNA-binding domain fusion的yeast cells進行mating,最後再進行 nutritional selection,所有在含有ORF-DA及ORF-BA的酵母菌細胞可以在difficient medium存活,並且經過重複的篩選以減少錯誤,再進行cDNA序列分析。
。
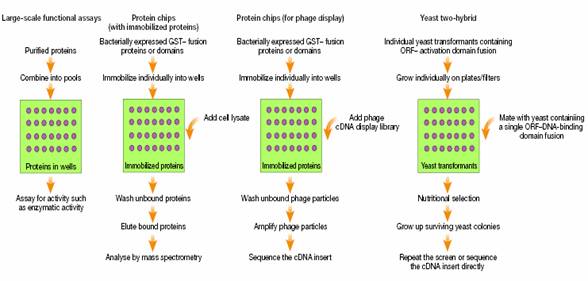
圖5:利用將某些蛋白質(如:GST-fusion proteins 或domains)固定在蛋白質晶片上,外加任何的蛋白質,如果蛋白質和蛋白質之間有相互作用的話,便會結合在晶片上,經過沖洗之後並不會掉下來,之後再利用mass spectrometry或Cdna序列分析,來鑑定蛋白質(2)。
蛋白質體學是運用2-DE、MS以及bioinformatics analysis的一項研究領域,它在訊息傳導的分析(signal transduction analysis)、心臟病(cardiopathy)、藥物動力學分析(drug action analysis)、機神分裂症的的病因、檢驗、解毒方法的分析(toxicology pathogenesis of schizophrenia)以及剪接體的新組合成份的鑑定(identification of novel components of splicesome)等方面提供了便利及眾多有用的訊息,但蛋白質體學並非萬能的工具,它仍需要基礎研究作為背景才能有所發揮,否則蛋白質體學則無用武之地。
1、Soren Naaby-Hansen, Michael D.Waterfield and Rainer Cramer, Proteomics-post-genomic cartography to understand gene function Trends in Pharmacological Sciences (2001) Vol .22:376-384
2、Akhilesh Pandey , Matthias Mann , Proteomics to study gene and genomes. Nature (2000) Vol 405: 837-846
3、Han-Jia Lin , Ching-Wei Luo ,Yee-Hsiung Chen , Localization of the transglutaminase cross-linking site in SVS III, a novel glycoprotein secreted from mouse seminal vesicle. J. Biol. Chem.(2002) Vol. 277: 3632-3639