蛋白質的交互作用
整理:施純如
在生物體中,蛋白質會彼此結合在一起,形成複合體,執行特殊的功能,這就是「蛋白質的交互作用」。功能單位有大有小,大如核糖體(ribosome)由30個以上的單位構成,小如轉錄因子(transcription factor)僅由2個組成。生化學家發現,幾乎所有的蛋白質都會與其它的蛋白質結合,或是透過不結合的方式,達到交互作用的目的,這種現象出現在高等動物(哺乳類)的頻率又高於低等的動物。因此,要了解細胞如何系統性運作之前,必須先了解蛋白質複合體如何共同執行功能;要知道蛋白質群體怎樣執行功能,就要先定義出這群複合體的組成。
欲明瞭群體中某蛋白質的功能,其線索在於結合的成員中,已經知曉功能的成員身上。例如,已知許多訊息傳導的蛋白激酶複合體(protein kinase signaling complexes),是由激酶(kinase,將磷酸根從ATP轉移到受體的酶)、磷酸酶(phosphatase,催化磷酸根由一化合物轉移到另一化合物上)、以及調控蛋白(regulatory protein)組成,其中激酶和磷酸酶的部份,可藉由分析它們的催化中心(catalytic domains),得知它們的基本生化功能,由此推斷複合體中的其它蛋白質,應屬於調控性質。
探測細胞層級的蛋白質結合,一般使用兩種實驗方法,一種是免疫沉澱(immunoprecipitation),如下圖:
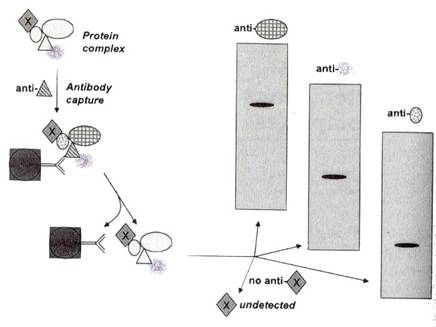
先以蛋白複合體其中一種的抗體與其作用(在圖中,就是三角形蛋白質的抗體),抓下這個複合體後,以SDS-PAGE和Western blot的方式將蛋白質轉印到膜上,最後用橢圓形、霧狀、圓形等三種蛋白質的抗體偵測,如果都有反應,就證明了三角形、橢圓形、霧狀和圓形等三種蛋白質的確會形成複合體;這個方法的先決條件是,必須有這些蛋白質的抗體,否則像圖中的X蛋白質,雖然也是複合體的一部份,但因沒有抗體,所以無法被測出。
另一種方法稱為「yeast two-hybrid system」,以間接的方式測得兩種蛋白質的交互作用,如下圖:
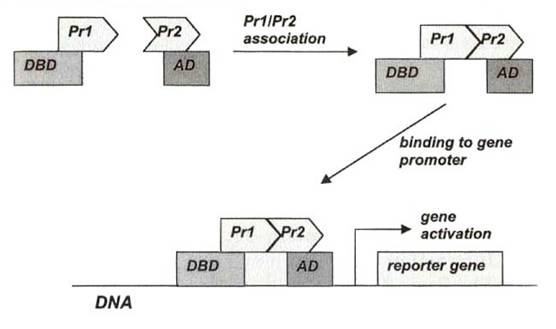
Pr1和Pr2是我們所感興趣的蛋白質,Pr1和Pr2是這兩種蛋白質的基因,將兩種基因與轉錄因子(transcription factor)融合(fusion),結合在啟動子(promoter)上,表現於酵母菌(yeast)中。DBD代表與DNA結合的部份,(DNA-binding domain),AD是活化的部份(activation domain),唯有兩種蛋白質彼此結合,形成完整的轉錄因子,它們所啟動的reporter gene才能表現;換句話說,是依reporter gene的產物是否表現,來推論Pr1和Pr2是否有交互作用。因為這是一種間接的方法,實驗過程中可能遭遇到其它的問題而影響結果,像是融合的基因產物無法到達細胞核,或是無法在酵母菌中表現。
直接以質譜儀作為分析工具,提供了一種探測蛋白質複合體組成的新方法,下面是簡單的流程圖:
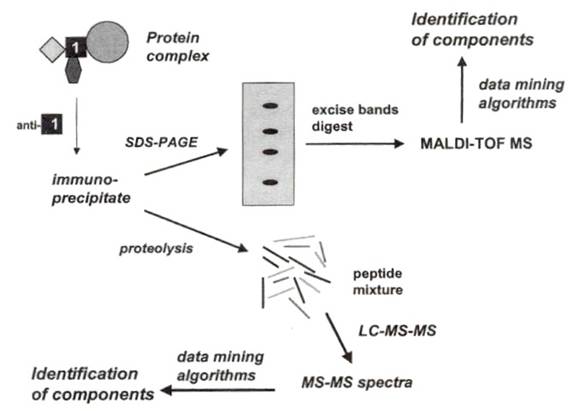
1代表我們所感興趣的蛋白質,與其它未知的蛋白質結合在一起形成複合體。將細胞萃取液以1的抗體作用,抓下蛋白質1,以及和它結合的物質(免疫沉澱),之後有兩種方法可以分析它,一種是電泳SDS-PAGE,染色後挑出蛋白質色帶(protein band),使用酵素分解它,最後用MALDI-TOF MS分析,藉由peptide mass fingerprinting algorithms 知道組成;另外,也可將膠體色帶中的peptide用LC-MS-MS得到MS-MS光譜(MS-MS spectra),比對database得到序列。使用SDS-PAGE可能面對的問題是,過程中會有蛋白質的流失,如分解膠體中蛋白質,分解得不完全,或是peptides從膠體分離出來的時候,分離得不完全。
除了電泳分離,還有一種方法,就是先用酵素分解,再用MALDI-TOF MS或LC-MS-MS分析,跳過電泳的步驟。如果複合體含的蛋白質在三個或三個以下,用MALDI-TOF分析是直接又簡單的方式,複合體的成員愈多,用MALDI-TOF spectra定蛋白質的難度就愈高,因此複雜的peptides混合物,通常選用LC-MS-MS spectra。
以抗體進行免疫沉澱(immunoprecipitation),是分離蛋白質複合體實驗中最常用到的方法。這個方法要能成功,首先要能取得適當的抗體,也就是能專一性抓到目標蛋白質,但在結合的同時,不影響目標蛋白與複合體其它成員的結合,因此並不是所有的抗體都適用於免疫沉澱。下面是免疫沉澱的簡圖:
在實際操作上,可以將抗體與當作支持物的「珠子」(beads)結合,再進行免疫沉澱,然後以1M的acetic acid這類的試劑進行短暫的變性作用(denaturant),使複合體與抗體分開,抗體以離心或過濾的方式回收,蛋白質複合體則進行酵素分解,送入MS分析。為了證明我們得到的蛋白質複合體,並不是在免疫沉澱過程中的人工疏失產物,還必須進行一些確認的工作,例如選用複合體中另一成員的抗體,進行免疫沉澱,同樣分離複合體、酵素分解、MS分析,比對兩種免疫沉澱結果是否相符。
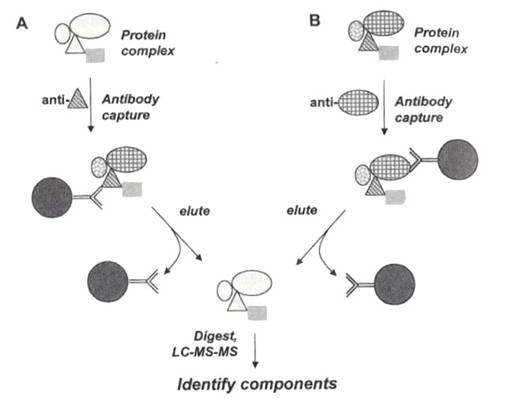
一種不使用抗體抓蛋白質複合體的方法,稱為「bait」。將目標蛋白質固定在固體支持物上,如下圖A:
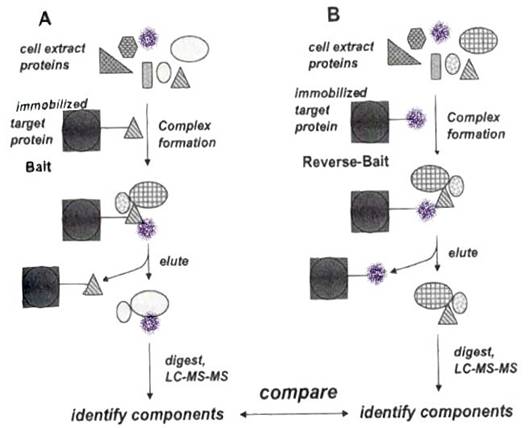
有許多方式可以把目標蛋白質連於beads,或是其它類似的支持物上。舉例來說,將蛋白質與活化的支持物(如epoxyalkylSepharose)作用一段時間,epoxyalkylSepharose會與蛋白質上親核性(nucleophilic)的-NH2(amine)團基,或是硫基(thiol group)共價鍵結;如果在蛋白質上標定His-tag或FLAG-tag sequence,使用鎳樹脂(nickel resins)或抗FLAG的抗體就能抓住此蛋白質。
把和bead結合的蛋白質與細胞萃取液,或是其它種類的蛋白質混合物一起培養,這些蛋白質形成的複合體會與beads結合在一起,將這一群體以離心或過濾的方式收集下來,經酵素水解和MS分析,便能得到複合體的蛋白質資料。
使用「bait」的優點有兩個:其一,不使用抗體,不干擾MS的分析;其二,不必擔心抗體是否能辨識複合體中的目標蛋白質。不可否認的,與支持物結合的目標蛋白,可能會影響它和複合體其它成員原本的結合力,例如它是以N端與支持物及其它成員結合的話,則兩邊的結合力會彼此干擾而減弱。Bait法的另一個缺點是,要讓目標蛋白質和支持物結合,比起單純購買抗體,所費的時間和金錢或許更多、更困難。
要確定bait法的正確性,可用「reverse bait」法,如圖B顯示。由bait實驗得到的結果,推知蛋白質複合體的組成後,我們挑選另一種蛋白質結合在bead上,重覆bait實驗,比對MS分析的結果看是不是相同。使用reverse bait法也許還能找到原本沒發現的複合體成員。
蛋白質除了能跟蛋白質結合外,它也能跟核酸上特殊的序列結合,轉錄因子(transcription factor)啟動子(promoter)的結合就是一例。研究蛋白質與核酸結合的方法,最常用的就是electrophoretic mobility shift assay,簡稱EMSA,如下圖所示:
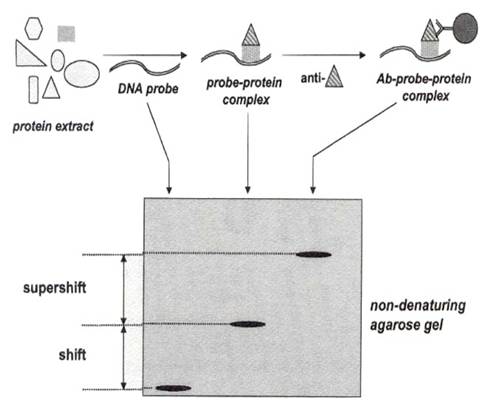
將一段含有欲探測序列的寡聚核苷酸探針(oligonucleotide probe)以32P標定,與含有欲測蛋白質的細胞液(cell lysate)共同培養一段時間後,混合物以不變性的狀態(nondenaturing condition)跑洋菜膠電泳,未與蛋白質形成複合體的oligonucleotide 在膠體中移動得較快;反之,標定的、與蛋白質結合的oligonucleotide移動得較慢,由膠體上可以看出兩者的差異(圖中標示的「shift」)。
要了解與核酸酸結合的是哪些蛋白質,可以在電泳之前,先以抗體作用一段時間,如果此抗體確實與複合體結合,這個群體在膠體中的移動速度,又會慢於單純的核酸-蛋白質複合體,如圖中標示的「supershift」,這樣就能確定複合體其中一種蛋白質成份,在這裡,抗體的選用就很重要了。
切下shift和supershift的band,其中的蛋白質用酵素水解、MS分析,便能得知它的組成,但是得到的蛋白質,產量通常十分稀少,因此往往要重覆實驗數次,把得到的蛋白質一起分析。
除了傳統的EMSA實驗,我們也可以改良之前描述過的「bait」法,測試蛋白質-核酸的結合。將oligonucleotide與支持物結合,如下圖:
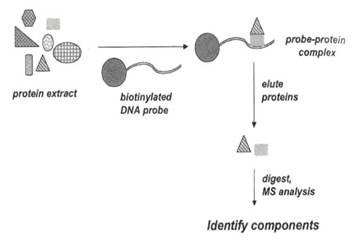
把oligonucleotide用生物素(biotin)標定,它能抓到會與它結合的蛋白質,連帶抓到此蛋白質形成的複合體,整個群體因為有biotin,所以能被帶有avidine的bead結合,非特異性結合的蛋白質,藉由洗脫的步驟去除,特異性結合的蛋白質便能純化出來,以酵素水解、MS 分析。
下圖是一連串以bait和reverse bait 方法探知「protein-interaction network」的流程:
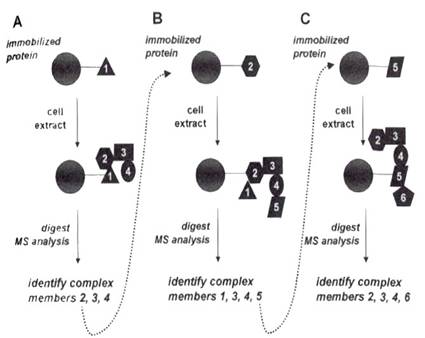
A:以protein 1為起點,讓它與bead結合,將這個餌(bait)與細胞萃取液作用,酵素水解、MS分析,得知其它組成包括protein2、protein3和protein4。
B:Reverse-bait,方法同A,但把一開始結合在bead上的「餌」換成protein2,得知其它組成包括protein1、protein3、protein4,以及新發現的protein5。
C:把「餌」換成protein5,經過相同的實驗步驟,以MS分析的時候,找到了protein2、protein3、protein4,還有另一個新組成protein6。
這樣的分析可以一直循環,以確認組成的完整性。當然,要把蛋白質餌與支持物結合在一起,是相當花時間的,因此若有適合的抗體,也可以選用免疫沉澱的方式交替使用。
Daniel C. Liebler Introduction to Proteomics-Tools for the New Bioloogy p151~164
Humana Press Inc. (2002)