等電點沉澱法
所有的氨基酸均為兩性物質,亦即它們至少含有一個酸性基(carboxyl)及一個鹼性基(α-amino)。這些可游離的團基在pH變化時,因釋出或接受質子而可充當弱酸或弱鹼,其離子化特性與其它物質一樣遵守Henderson-Hasselbalch方程式:
pH = pKa + log10 [未質子化的形式(鹼)] / [已質子化的形式(酸)]
即
pH = pKa + log [A-] / [HA]
氨基酸可以三種形式存在,即正電荷(cation)、兩性離子(zwitterion)或雙極性離子(dipolar ion)及負電荷(anion)等三種,若在酸性溶液中帶正電荷,則在鹼性溶液中帶負電荷。若氨基酸在某一pH值下其淨電荷為0,且在電場中不移動時,稱此pH值為它的pI值(等電點)。因為淨電荷為零,淨電斥力不存在的緣故,大部份蛋白質於等電點的pH值下,其溶解度最小。相反的,當溶液的pH值低於或高於pI,所有蛋白質分子所帶淨電荷必為同號,彼此之間有相斥力,不會凝結。所以,將pH調到等電點的大小,則大部份的蛋白質將會沉澱,這種現象可以應用於估算某蛋白質的等電點;另一方面,也可以應用在電泳,達到分離蛋白質混合物的目的,這種方法稱為等電聚焦電泳(isoelectric focusing),簡稱為IEF。
Isoelectric focusing是在具有pH梯度環境中進行的電泳。將蛋白質置於不同pH梯度的膠體進行電泳,它會朝著與自身所帶電性相反的電極方向移動,直到抵達與等電點(pI)相同的pH值處才停止,如果它移到別的pH處,會因為帶電而再度移動回和它pI相符的pH處,這就是為什麼此法被稱為「focusing」的原因。
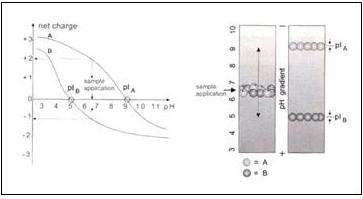
上圖左以A,B兩種蛋白質為例,作出它們的淨電荷曲線圖,橫軸代表環境中的pH值,縱軸代表蛋白質的淨電荷,在pH6.5時,A帶有兩個正電荷,B帶有一個負電荷。使A與B的淨電荷為0的pH值分別為pH9和pH5,這就是它們的「等電點」。
上圖右是A與B在isoelectric focusing gel中移動的情況,一開始將A與B加在膠體pH值為7的地方,之後兩種蛋白質會移往與各自等電點相符的pH處,A是pH9,B是pH5。
一般形成Ph梯度有2種方法:
(1)人工pH梯度,這是在電場存在下,用兩個不同pH的緩衝液互相擴散平衡,在其混合區間即形成pH梯度,但這種pH梯度受緩衝液離子電遷移和擴散的影響,因而pH很不穩定,常見於製備柱電泳。
(2)自然pH梯度,這是60年代初,Svensson(後來改姓Rilbe)引入的概念,並利用數學推導,為等電聚焦奠定了理論基礎。自然pH梯度是利用一系列兩性電解質載體在電場作用下,按各自pI形成從陽極到陰極逐漸增加的平滑和連續的pH梯度,此pH梯度進程取決於各種兩性電解質的pI、濃度和緩衝性質。因為擴散和電移動所引起物質移動處於動態平衡,所以在防止對流的情況下,只要有電流存在就可保持穩定的pH梯度。在此pH梯度中,各種蛋白質遷移到各自的pI處,而得到分離。PH梯度的形成是IEF的關鍵,Svensson設想理想的兩性電解質載體應具備下列條件:
(1)易溶於水,在pI處應有足夠的緩衝能力,形成穩定的pH梯度,不至被蛋白質或其他兩性電解質改變pH梯度。
(2)在pI處應有良好的導電及相同的電導係數,以保持均勻的電場。
(3)分子量小,可通過透析或分子篩法除去,便於與生物大分子分開。
(4)化學性能穩定,與被分離物不起化學作用,也無變性作用,其化學組成
不同於蛋白質。
要製作含不同pH梯度的膠體,首先必須準備酸性與鹼性的acrylamide溶液,利用gradient maker將兩者緩緩以不同比例混合,如下圖所示:
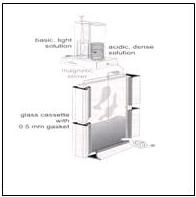
酸和鹼的溶液,加入的量會被自動控制,因為調配酸性溶液時,將其比重加大,故一開始它會沉在下面,隨著鹼性溶液加入,pH慢慢提高,整片膠體便產生pH的梯度。
目前,兩性電解質載體商品由於生產廠家不同,合成方式各異,而有不同的商品名稱,如Ampholine(LKB公司)、Servalyte(Serva公司)、Pharmalyte(Pharmacia公司)。IEF-PAGE分離蛋白質並測定pI時可先選用pI 3-10的兩性電解質載體,以及同一範圍的標準pI蛋白質,將其與樣品同時電泳,固定染色後,就可以pH為縱軸,具陰極遷移距離(cm)為橫軸,做出pH梯度標準曲線,根據染後蛋白質遷移距離,可推知其pI。為進一步精確測定未知物的pI,還可選擇較窄範圍的兩性電解質進行電泳,以提高分辨率,得到更準確的pI。如實驗時,無標準pI蛋白質作為標定依據,則電泳後立即用表面微電極每隔0.5公分直接測定膠板的pH值,製作pH梯度曲線,染色後根據遷移距離推知某種蛋白質的pI。
誠如前面所提,梯度膠體目前廠商已上市,一般的pH範圍是3到10,針對所要觀察蛋白質的性質,也可以選擇3到12,4到7,6到11,6到9的特製凝膠。買來的膠條在使用前需進行「復水」(rehydration)的動作,復水用的緩衝液成份視膠體而定,時間從6小時到overnight不等。整個步驟如下:
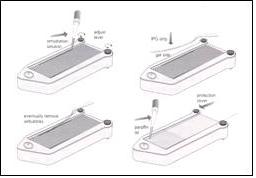
先在下方的「reswelling tray」中滴入復水用的溶液,再將膠條放入,小心去除氣泡,使膠體與溶液均勻接觸,覆上一薄層的蠟油防止水份蒸乾,最後蓋上蓋子。
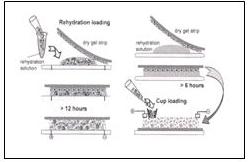
樣品也可以在膠條復水的時候一起加入,方法如下:可以選擇復水之前加入(左),也可以在復水6小時後加入(右),前者的復水時間要超過12小時。
將加好樣品的膠條架上電泳槽,即可開始進行電泳:
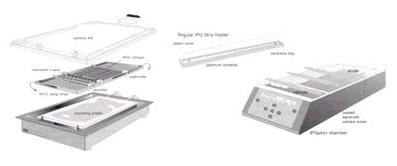
一個電泳槽可以放置數個膠條,電源供應器能設定不同條件,最高提供8000伏特的電壓,並附有冷卻系統,避免高電壓帶來的高溫影響膠體的品質。 雖然電泳槽的設計,可以讓長短不同的膠條一起進行電泳,但仍不建議將大小、pH梯度不同的膠體一起進行實驗。
當用PAA(polyacrylamide)或瓊脂醣(agar)作為穩定介質時,有時最後測得pH梯度常與兩性電解質載體標明的pH範圍有差別,這可能與電內滲有關。因此IEF必須使用無電內滲的高純度穩定介質。在IEF-PAGE中,丙烯醯胺純度極為重要,最近Pharmacia公司推出Amberlite MB-6或採用類似H+和OH-混合樹脂除去丙烯酸,其效果較再結晶法更佳。在IEF-PAGE中,凝膠只是一種抗對流支持介質,並無分子篩作用,因此凝膠濃度的選擇只要形成的孔徑有利於樣品分子移動就行,一般用5%或7%均可。
兩性電解質載體是IEF-PAGE中最關鍵的試劑,直接影響pH梯度的形成,以及蛋白質的聚焦。因此,要選用優質兩性電解質載體,在凝膠中,其終濃度一般為1-2%。PH梯度的線性依賴於兩性電解質的性質,選擇哪種pH梯度範圍的兩性電解質載體,則與被分離蛋白質的pI有關。
應選擇在電極上不產生易揮發物的液體,作為電極緩衝液,陰陽電極溶液的作用,是避免樣品及兩性電解質載體在陰極還原,或在陽極氧化,其pH應比形成pH梯度的陰極略高,比陽極略低。值得注意的是,不同廠商合成兩性電解質方法不同,應根據說明書選用有關電極溶液。
在採用AP催化化學聚合時,為防止氧分子存在影響聚合,因此加AP前應將溶液抽氣。此催化系統在鹼性條件下容易聚合,在酸性條件下(pH<5)凝膠聚合比較困難,這可能是在酸性條件下,AP不能充分產生出氧原子,使單體成為游離基,因而阻礙凝膠聚合,可在凝膠中加入1%的AgNO3促使凝膠聚合。凝膠聚合後,為防止酵素活性降低,在加樣前進行15-30 min欲電泳,然後將樣品放在其pI附近。
在pH 3-10、中性及鹼性pH條件下,加入TEMED可加速凝膠聚合,但在pH<5時則加速作用較少。TEMED本身為鹼性物質,在pH 4.5以上能擴展PAA凝膠鹼性端pH梯度,其擴增幅度與TEMED加入量有關。實驗表明在20 ml凝膠溶液中,加入50 ul TEMED,在距陰極0.3 mm處可擴展0.7 pH;加100 ul TEMED在陰極處能擴展1.3 pH,也就是說pH 3.5-9.5的兩性電解質載體可擴展到pH 3.5-11的梯度範圍。因此加入TEMED對鹼性蛋白質的分析極為有利,可用於分離細胞色素c、組蛋白等。
實驗證實,鹽離子可干擾pH梯度形成,並使區帶扭曲。例如,將L-胺基酸氧化酵素分別溶於不同濃度的NaCl, Tris-HCl緩衝液中,從IEF-PAGE染色圖譜可看出,高濃度及低濃度Tris-HCl緩衝液對pH梯度形成影響較小,而NaCl及磷酸鈉緩衝液干擾pH梯度形成並使區帶扭曲。為了防止上述影響,進行IEF-PAGE時,樣品應透析或用Sephadex G-25脫鹽,也可將樣品溶解在水,或是低鹽緩衝液中,使其充分溶解,以免不溶小顆粒引起拖尾。但某些蛋白質在等電點附近,或水溶液及低鹽溶液中,溶解度較低,則可在樣品中加入兩性電解質,如加入1%甘胺酸或對1%甘胺酸透析,雖然甘胺酸是兩性電解質,但不影響pH梯度的形成,可利用其在溶液中的偶極矩作用增加蛋白質的溶解性;也可將樣品溶解在含有2%兩性電解質載體中,因他含有可反應的胺基,並能除去氰酸鹽,此時樣品最好加在經預聚焦的凝膠板的陽極側,因在pH 5以下,既沒有氰酸鹽也沒有胺基甲醯化存在。此外,還可在樣品及凝膠溶液中加入分離子去污劑如Tween 80, Triton X-100, Nonide P-40等或加入相同濃度的尿素(4 M),為防止氰酸鹽引起蛋白質的胺甲醯化,含有尿素的樣品及凝膠板只能當天使用。
加樣樣取決於樣品中蛋白質的種類、數目及檢測方法的靈敏度。如用Coomassie Blue R250染色,加樣量可為50-150 ug;若用銀染色,加樣量可減少到1 ug。一般樣品濃度以0.5-3 mg/ml為宜,最適加樣體積為10-30 ul。如樣品很濃,可直接在凝膠表面加2-5 ul;如樣品很稀,可加樣300 ul。將其放在一特製的塑料小框中,或用一塊泡沫塑料及高品質的濾紙,也可用擦鏡紙吸取樣品放在凝膠表面。由於IEF-PAGE是按蛋白質的pI分離,電泳後各種蛋白質被濃縮並停留在其pI處。電泳後均可得到同樣的結果。值得指出的是:對不穩定的樣品可先將凝膠進行15-30 min預電泳,使pH梯度形成,然後將樣品放再靠近pI的位置以縮短電泳的時間,但不要將樣品正好加在pI處和緊靠陽、陰極的膠面上,以免引起蛋白質變性造成條帶扭曲。一般加樣電泳半小時後,取出加樣濾紙以免引起拖尾現象。
電功率是電流與電壓的乘積。在IEF電泳中,隨著樣品的遷移越接近pI時,電流則越來越小。為使各成分能更好地分離,要保持一定的電功率,就應不斷增加電壓,電壓增高可縮短pH梯度形成,和蛋白質分離所需的時間,但過高的電壓會使凝膠板局部範圍,由於低傳導性和高阻抗而過熱、燒壞,為此,在電泳過程中,應通冷卻水,水溫以4-10℃為宜,流量5-10 l/min。避免使用過低的溫度,以免冷凝水滴形成。超薄板(0.5 mm)TFE分辨率高就是因為易冷卻。
IEF-PAGE時間與電功率取決於多種因素、如聚丙烯醯胺的品質、AP和TEMED用量、膠板厚薄、兩性電解質載體的導電性和pH範圍。窄pH範圍電泳時間比寬pH範圍時間長,這是因為在窄pH範圍蛋白質遷移接近pI,帶電荷少,故遷移慢。為了提高分辨率,就要增加電壓,縮短電泳時間,防止生物活性喪失。對未知樣品可進行不同電壓、時間的電泳實驗,此時可將有色蛋白質(如血紅素)作為標誌,將其放在不同位置,當聚焦帶遷移到同一位置時,說明已達到穩態,一般寬pH範圍電泳時間以1.5-2 h為宜。
IEF-PAGE操作簡單,只要一般電泳設備就可進行,電泳時間短、分別率高、應用範圍廣,可用於分離蛋白質及測定pI,也可用於臨床鑑別診斷、農業、食品研究及動物分類等各種領域。隨著其他技術的不斷改進,等電聚焦電泳也不斷充實完善,從柱電泳發展到垂直板,又進而發展到超薄型水平板等,還可與其他技術或SDS-PAGE結合,進一步提高靈敏度與分辨率。
參考資料:
Reiner Westermeier, Tom Naven
Proteomics in Practice- A Laboratory Manual of Proteome Analysis
p187~p197 WILEY-VCH (2002)
整理: 施純如
李建武等合編 生物化學實驗原理和方法 P155~163 藝軒圖書出版社 (1999)
整理: 黃信明