蛋 白 質 分 離
口述:吳啟裕 博士
整理:張書菀、吳皓芝、吳辰宇、梁曉政
二、蛋白質體學的表現(Expression Proteomic)
四、蛋白質的準備(Protein Sample Preparation)
五、蛋白質的分群(Protein Sample Prefractionation)
最近數年間,蛋白質的分離、定量、定性之技術已大幅度的改良。而各項分離技術之間的結合、運用,使蛋白質能更有效的被分離,而參與後續的各項研究程序。由於蛋白質分子之間不同的特性(例如:分子大小、pI質),可發展出多維(multidimensional approach)的分離技術,例如:2-D electrophoreis就是以蛋白質的電荷(第一維)與分子量大小(第二維),已達分離的目的。然而各種分離技術均有優缺點,因此選擇適當的方法是分離的關鍵。
什麼是蛋白質體學呢?Yates定義為:紀述並且分析蛋白質、蛋白質間的交互作用(protein interaction)、蛋白質之修飾(protein modification)的科學學科。簡單的來說,就是研究來至於細胞、組織或個體中的蛋白質,並以分子的層級來說明牽連蛋白質的各項細胞反應、過程。在下面的報告裡裡除了固有的蛋白體學上的各項技術之外,還有新穎的的分離(separation)、檢定(detection)與定量(quantification)將被討論於其中。蛋白質體(proteome)在研究上比基因體(genome)就困難,因不同形式的細胞具有不同的蛋白質體。
因為一細胞中的蛋白質體並不是單一且固定的,它結合基因體、當下的環境、甚至該細胞的演化歷史,在人類的細胞中,約有20000蛋白質其是在特殊類型的細胞中才被表現出,又其表現出來的蛋白質濃度在不同時刻均不相同,因此更加深了蛋白質體在分析與研究上的複雜性。在2-D electrophoreis的分離技術中,蛋白質在膠體中的含量(約50-75%)因為含量的不足導致難以利用2-D電泳分析來檢測,因此在樣本的製備(preparation)、濃度(concentration)、分離(separation)及檢測(detection)等方式均需要重新研發,以解決2-D electrophoreis的缺點。
二、蛋白質體學的表現(Expression Proteomic)
1. Gel base:
*優點—1.唯一一種能一次同時分離2000個蛋白質的方法。
2.能得到為影像的實驗結果。
3.從實驗結果能得知sample protein的pI(等電點/isoelectric point)及M.W(分子量/molecular weight)所以能知此protein是否有isoform及post-transcription modification。
*缺點—1.膜蛋白(hydrophobic)及M.W較小或較大的蛋白質(小於16kD、大於150kD)在電泳結果中較不易看到,此外,pI較偏酸性的也不易看到。
2.實驗結果所得的樣品回收率較低。
3.需要花很多時間做sample preparation才能得到較可信而準確的實驗結果。
4.無法自動化à最大缺點。
2. LC base:
缺點:MDLC在做時,是將蛋白質都切成peptide,因此會容易遺失掉一些訊息
優點:好處是靈敏度較高,而且可以自動化
由於不可能將一個生物所有的蛋白質全部分離出,再全部一起去分析,這樣會無從下手,照成不必要的浪費,因此在實驗之前要先想清楚實驗目的為何,再根據所要做的實驗、要分析什麼樣的蛋白質,去決定如何做樣本前處理,處理完後再決定要用哪種方法去分離出所要的蛋白質。找出哪個方法對自己的實驗才是最好的方法。
流程:
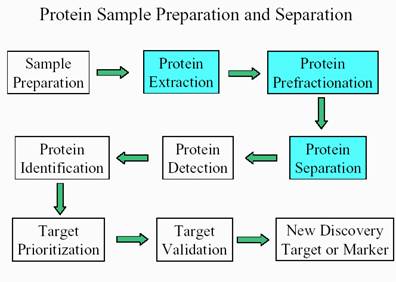
![]() 拿到一個樣品,該如何做蛋白質的萃取?要配合適當的樣品前處理。
拿到一個樣品,該如何做蛋白質的萃取?要配合適當的樣品前處理。
![]() 萃取完之後,要做分組的動作(prefractionation),將一次所要分析蛋白質的數目縮小,以方便分析
萃取完之後,要做分組的動作(prefractionation),將一次所要分析蛋白質的數目縮小,以方便分析
![]() 蛋白質的分離
蛋白質的分離
四、蛋白質的準備(Protein Sample Preparation)
1. Protein Extraction
(1)萃取的過程主要要注意兩件事:
1. To completely extract the total protein:將所有要的蛋白質完全萃取出來,如未萃取完全可能會造成極大的誤差,在做比對工作時會造成誤判。
2. To remove non-proteins:如何將非蛋白質得物質完全移除掉
(2)萃取時需考量的因素
1.使用的樣品是細胞或著是堅硬的組織
2. 是否需要做分組的動作:由大範圍縮減成小範圍以利分析
3. 哪些干擾的物質需要去排除?要移除的東西不見得不是蛋白質,有可能要移除掉大量存在的蛋白(如血液中的albumin、immunoglobin,量太多照成大量干擾)
4. 要部分分離或是全部分離
(3)應用到的方法共有:
a. Cell or tissue sample distruption:打破的方法。
b. Protein precipitation:沉澱的方法,如利用TCA(trichloroacetic acid)沉澱,把要的蛋白質沉澱出來,不要的留在溶液中。
c. Protein Solubilization:如何把想要的蛋白質溶解,如一些膜蛋白為hydrophobic,非常難溶解。
d. Protease activity Inhibition:不能用太強的detergent,也不能利用TCA這種強烈的或化學物質,可能會破壞蛋白質的三度空間結構而失去活性。
e. Removal of non-protein,非蛋白質的部分包括:
- nucleic acids
- lipids
- salts, buffers, ionic small molecules
- insoluble material
a.打破細胞的方法:
1. Free-thaw or osmotic lysis:放入液態氮內急速冷凍,再放入37℃ incubator,讓細胞處於忽冷忽熱的環境,比較軟的細胞會因此而破掉,如果是只要分離細胞核的話就可以利用這種方法。
2. Detergent lysis:可以用一些detergent溶掉細胞膜,使細胞內的物質流出。
3. Sonication:超音波震盪,比較強烈的方法,會打爛所有的胞器,在想看細胞內所有的protein時使用,通常是事先未知我們要看的蛋白質為在何處。
4. Enzymatic lysis:比較溫和的方法,只要去破壞細胞的某一些結構。可利用 pectinase, cellulasae, lysozyme or lyticase™, 例如:只想要移除細胞壁時可利用cellulase。
5. French pressure cell:高壓的方法,通常用在有細胞壁的微生物
6. Grinding(motar and pestle):用研磨的,通常用在比較堅硬的組織
7. Machanical homogenization:均質器,也屬於研磨的方法,通常用在比較堅硬的組織
※ 最好的方法:是要根據所要研究的對象去判斷
b.沉澱的方法:
1. Ammonium sulphate(salting out):將大量ammonium sulphate加入蛋白質溶液中,隨著鹽類濃度的增加,與蛋白質競爭水分子,蛋白質的hydophobic suface會曝露出來,因為hydrophobic interaction,蛋白質彼此間會聚集而被沉澱出來。用此法容易保留蛋白質活性,但是會殘留大量鹽類,很難去鹽,而且有時候用硫酸銨沉澱效果不是很好
2. TCA(trichloroacetic acid) precipitation:使蛋白質溶液的pH改變,而達到蛋白質的等電點,使蛋白質沉澱下來。沉澱效果好,可以大量去做,但是強烈的藥物容易破壞蛋白質的三級結構,而使之喪失活性。
3. Acetone and/or ethanol:蛋白質在含有不同濃度有機溶劑的溶液中,會具有不同的溶解度。隨著加入有機溶劑濃度的增加,與蛋白質競爭水分子,使蛋白質曝露出charge group,因發生ionic interaction而聚集沉澱下來。
4. TCA plus acetone
c. 蛋白質的溶解(Protein solubilization)
1. Urea(8-9.8 M), or 7 M urea / 2M thiourea:替蛋白質作一些unfolding的動作,希望蛋白質不會全部糾結在一起,這樣做IEF時蛋白質才會停在正確pI的位置,不會有一些負電荷包埋在裡面的情形
2. Detergent(CHAPS [3-(3-cholamidopropyl)dimethylammonio]-1- propanesulfonate),…):有一些蛋白質在緩衝液或水中不好溶,因此會加入一些detergent幫助溶解,但是在做IEF時會通很高的電壓,所以用的detergent一定要是電中性,不然會造成很強的電流,使蛋白質變性,更嚴重的是會造成整個電路的短路。CHAPS是一種zwitterionic detergent,是目前很常用的detergent。SDS不適用,SDS帶負電。
3. Reductant(DTT, DTE, TBP):去掉胺機酸的Cysteine的雙硫腱,DTT(dithiothreitol)可以用來還原雙硫鍵,以利作蛋白質電泳分析,在溶液中含有低溶度(1mM)dithiothreitol時,有避免蛋白質氧化的作用。DTE(dithioerythritol)在pH大於8會帶負電,會對IEF有不良影響。新開發出來的還原劑TBP(tributyl phosphine)不帶電,不受電場及pH的影響,同時對於膜蛋白跑2D也比較好。
4. Carrier ampholyte(IPG buffer):用來做pH gradient
l Sonication can help solubilization:
樣品處理完,藥都加完,再做超音波震盪一下,可以增加蛋白質的溶解度,在做2-D electrophoresis有利蛋白質的分離。增加蛋白質的溶解度非常重要,因為在做IEF dimension時,是讓蛋白質停在等電點的位置,在等電點的時候,蛋白質溶解度最差,容易沉澱下來,一但沉殿,就卡在膠體上,之後去做2D second dimension時就不會在移動,最後就不會在膠體上看到蛋白質的正確位置。因此跑2D時問題通常都是出在IEF dimension,會有沉澱的問題以及雜質的問題。
l Sample can be heated only perior to addition of urea:
加urea之後不可以再加熱,因為urea加熱時會產生另外一種化學物質,形成雙鍵,修飾在蛋白質上,特別會修飾胺機酸lysine,使得trypsin 原本切胺機酸lycine C-terminal位置因為被urea修飾而造成不能切,使得要做蛋白質分析時,protein沒有辦法變成較小的peptide片段,做質譜儀分析的時候就不能精確的策到peptide片段。
d.Protease inhibitors:
在分析的蛋白質中難免會殘留一些protease在我們加入trypsin去作用這些蛋白質時,難免會先被這些protease破壞,因此一定要確定抑制住protease的活性。
1. 苯甲基磺醯化氟PMSF(phenylmethylsulfonyl fluoride):最常用的,用來抑制serine and cysteine protease的活性。具有毒性,在水溶液中可藉由DTT and 2-mercaptoethanol來去PMSF活性。
2. AEBSF(Aminoethylbenzylsulphonyl fluoride):也是用來抑制serine和cysteine protease,水溶性較好,毒性較PMSF低,但可能造成蛋白質彼此共價鍵互相作用,影響蛋白質的pI值。
3. EDTA:抑制matalloprotease。
4. Peptide protease inhibitors(leupeptin, pepstatin, aprotinin bestatin…etc):可用來抑制Serine-, cystine-, aspartyl-proteases, aminopeptidases,缺點是成本較高,且有可能出現在2D pattern中。
5. High pH:抑制大部分的protease。
e. Nucleic acid removal
核酸也是細胞中含量非常多的一部份,也要將之移除乾淨,移除可利用下列方法:
DNase I and Rnase A are commonly used:可利用酵素移除,但要注意DNase本身也是一個蛋白質,必須要注意加入是否會污染到sample。
Nucleases:Nucleases本身也是protein,因此也會受到urea的破壞,當在8M urea中就無法作用,所以必須在加入urea之前就加入。
DNase I :會出現在2D map中,因為本身也是蛋白質
Benzonase:同時具有DNase 及RNase的活性,可同時去掉DNA and RNA,目前很常用
Sonication works very well:有的人不喜歡用Nuclease,直接用超音波去振,然後用超高速離心將DNA沉澱下來,此方法可以避免可能污染的問題,也比較省錢
f.De-salting techniques
為何要去鹽?如果不去鹽,在跑2D or SDS的時候,因為鹽類也是導電物質,會造成導電度變大,產生大量的熱。
˙Dialysis:利用擴散的原理,很難百分之百去乾淨,有一點殘留上2D的還是有問題,電壓上不去。
˙Spin dialysis:利用過濾膜,然後去離心,離心力太大的話,連分子量很大的都過的去,回收率不好,因容易殘留在膜上。
˙Gel fitration:好處是去鹽可以去的很乾淨,但缺點是因為要流colum,當sample多的時候會耗掉非常多時間。
˙TCA 沉澱:可以很有效的去鹽,但是由於沉澱的東西非常緊密,sample非常難回溶,很耗時。
※注意:各種方法都有優缺點,沒有一種完美的方法。要考量所要研究的東西,實驗的目的去決定要應用哪一種方法或是配合使用。
五、蛋白質的分群(Protein Sample Prefractionation)
1. 前言
我們知道細胞中有分abundant protein和low abundant protein,對於要分析細胞中abundant protein可省去 ”prefractionation” 步驟而直接進行二維電泳觀察所求蛋白質;但若所要分析的蛋白質屬於細胞中的low abundant protein,則欲得到準確而可信的實驗結果就必須要作sample prefractionation,將大量的蛋白質作「分群」動作,以增加之後蛋白質分離(separation)的效率。
2. Sample prefractionation的目的在:
*To remove abundant proteins—可利用離心或不同蛋白質具不同溶解度的方式。
*To enrich low abundant proteins—可利用affinity column或antibody抓出所要的
protein。
*To reduce the protein complexity
以上目的的滿足點決定於:「進行二維電泳能分離的總量」,以及「要看的protein是否能被觀察到」。所以要根據所欲觀察protein的複雜度來決定以上因素的應用。同時,prefractionation後不見得每個protein都要單獨分開(95%以上純度)才能被identify,亦可藉由LC/MS/MS而鑑定出一個fraction中的protein。如:利用IEF(first dimention)àSDS PAGE(second dimention)àLC(third dimention),此種分離方式屬於multiple dimention。
3. 常用方法
一般而言,純化某種待測蛋白質的步驟是先將取得的細胞打破,均質化,從中萃取出蛋白質。先利用每種蛋白質溶解度不同,例如經由簡易的硫酸銨沉澱法(salting-out)將其粗分離,再依據所求蛋白質的物理(如:大小、比重)及化學性質(如:所帶電荷、極性、親和力)再將其作進一步細分。
(1)次細胞分劃(Subcellular Fractionation)—若所求蛋白質為細胞中某一胞器所特有的,則需先將該胞器分離出來。通常不同胞器可利用”differential centrifugation”或”equilibrium density-gradient centrifugation”等方式分離。
a.Differential-velocity centrifugation
原理:不同胞器其大小密度不同,所以可利用逐漸增加離心速度的方式將不同胞器分別分群地分離出來。
步驟:細胞均質化後,先以過濾或低速離心的方式,除去未破碎的細胞或細胞破片。而後再以約600g(約500 rpm)的轉速離心十分鐘,將細胞核沉澱分離。取上清液,加快離心速度至15000g,五分鐘後將mitochondria、chloroplasts、lysosome,peroxisomes沉澱,在將轉速增至100000六十分鐘,沉澱plasma membrane、microsomal fraction和large polyribosomes,轉速在增至300000g兩小時,沉澱ribosomal subunits和small polyribosomes,將上清液到掉,留下沉澱物,但是這些離心的轉速與時間是依你想要取得的胞器的不同而隨著改變。
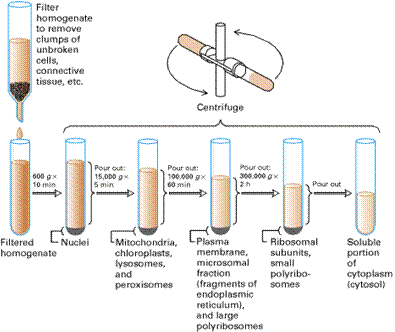
圖1、Differential-velocity centrifugation 大致流程圖:細胞均質化後,先將細胞碎片過濾掉,利用離心轉速的不同與時間的長短來分離細胞內各胞器(1)。
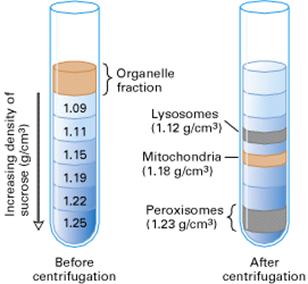
b.Equilibrium density-gradient centrifugation
分離效果較differential-velocity centrifugation更佳。
原理:依據不同胞器具有不同密度而將其分離。
步驟:離心管內具有無導電性的基質所形成的密度梯度,如:glycerol或sucrose。欲細分的organelle fraction置於上述物質的最上層,而後以高速(約40000 rpm)離心數小時,使在organelle fraction中的胞器能移動到和其本身密度相當的部分。

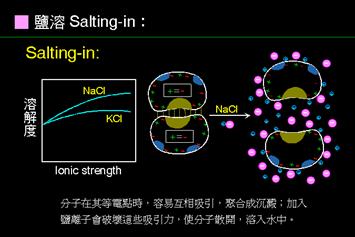
(2)利用不同溶解度(Differential solubilization)—利用蛋白質溶解度不同而將其分離。例如:鹽溶法(salting-in)。又如:對可溶於水的蛋白質可增加溶液中detergent(利用urea或thiourea)濃度增加其溶解度。
a.鹽溶:
原理:鹽類溶解所解離出的大量正負離子阻隔蛋白質本身所帶電荷的相互吸引作用,而增加蛋白質在溶液中的溶解度。
。

b.ReadyPrep Swquential Extraction Kit:
利用不同蛋白質在不同溶液中具有不同溶解度的性質,將其分別分離出來。如下圖4為例,一開始用Tris pH 9.5溶液溶解蛋白質,收下能溶於Tris的蛋白質。剩餘不溶的蛋白質再利用較強的detergent,如:Urea/CHAPS/Tris或Bio-Lytes/TBP溶液,使其溶解,進而收取更hydrophobic的protein,之後再利用更強的detergent,如:Urea/Thiourea/CHAPS或SB3-10/Tris/Bio-Lytes/TBP等萃取更加hydrophobic的protein。所以以此例而言total cell作二維電泳所得結果的圖是三片2D 膠體的影像相加的結果。但若不作prefractionation而直接以cell打破後取得的total protein做2D的圖,產生的一個spot可能包含數種蛋白質。

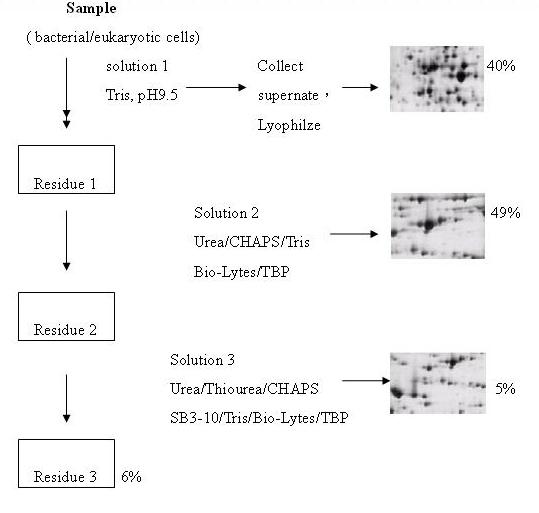
圖4、ReadyPrep Sequential Extraction Kit使用流程:利用不同的液體溶解不同的蛋白質
,將每一步驟所溶解的蛋白質去跑電泳,所得的結果才是總蛋白含量(4)。
(3)色層分析(Chromatography)—管柱層析法為色層分析法的衍生改良,能將粗萃取蛋白的純度提升至50~90%。一般而言,層析法所使用的實驗系統一定包含兩相:流動相及固定相。分離蛋白質所使用管柱層析法的固定相一般為大小均等,耐高壓且和蛋白質無專一性吸附的多醣類膠球,如:Sephadex(葡萄聚糖)及Sepharose(洋菜糖),而流動相則是含樣本蛋白質的緩衝液。常用的管柱層析法包括:Gel filtration、Ion exchange chromatography、Affinity chromatography等,原理分述如下。
a.膠體過濾法(Gel filtration):
固定相:內含小孔徑的膠球。
流動相:含樣本蛋白的緩衝液。
當樣本蛋白流經膠球時,小分子的會經由膠球孔徑進入膠球內,增加分子在管柱中移動路徑因而延長被分離出的時間。大分子物質因為無法進入膠球內所以能優先被分離出。
b.離子交換色層分析(Ion exchange chromatography):
固定相:表面帶有離子基團的膠球。
流動相:含樣本蛋白的緩衝液。
由於蛋白質表面具許多帶電基團,因此可藉由分子間作用力(相吸或相斥)不同而達到分離所求蛋白的結果。一般又可分為陽離子交換及陰離子交換兩種不同的層析法。
c.親和層析法(Affinity chromatography):
固定相:表面帶有特定親和基團的膠球。
流動相:含樣本蛋白的緩衝液。
由於某些分子彼此間具有結合專一性(如:抗原與其抗體、DNA兩股,等。)因此可利用此特性將我們要的蛋白質從混合物中分離出來。
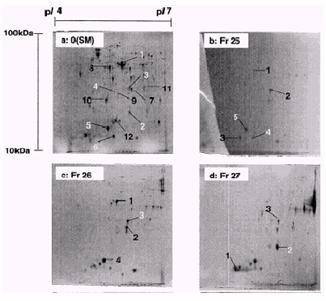
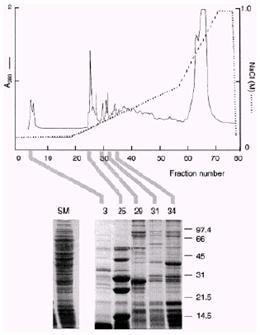
圖5、利用色層分析先將蛋白質分開,再進行2-D電泳分析,較蛋白質直接跑2-D
電泳分析的複雜度為低(5)。
(4)裝備式電泳(Preparative Electrophoresis)—用以分離大量的protein sample。
電泳是一種十分高解析度技術,可用於蛋白質的純化。然而,它有兩項重要的缺點。一為能分離蛋白質的量有限(limited amount of protein that can be loaded onto a gel);一為實驗後自凝膠( gel )中所能回收蛋白質產物的量有限,尤其是當gel已被固定且經過染色。
將蛋白質自gel中回收的方式通常有兩種。一是利用擴散( simple diffusion ),將切下的膠體塊浸泡於適當緩衝液中,讓其內含的protein自然擴散出來。這種方式的缺點是protein自然擴散的速度極為緩慢,且所得蛋白質會被過度稀釋,通常需再利用酸沉澱的方式才能取得;另一則是利用電泳溶離(electroelution)方式,將膠體塊置於一種商業化儀器中(commercially available apparatuses)內,或置於透析袋( dialysis bag) 中。利用通電的方式將不要的高分子溶液中的電解質和低分子的不純物移除。上述兩種方式的共同缺點為,protein回收產量極低。鑒於上述缺點,因此發展出一種商業化的裝備式電泳(preparative electrophoresis)儀器,將gel設計為圓柱形,且在其底部設計有一個密封的透析膜。蛋白質跑到膠體底部後會移動到膠體外,而利用該透析膜收集所得的蛋白質,收集到的proteins又可被分入monitor及fraction collectors中。
現在取代傳統的固態物質的製備式電泳有兩種儀器為Rotofor and Free flow electrophoresis。
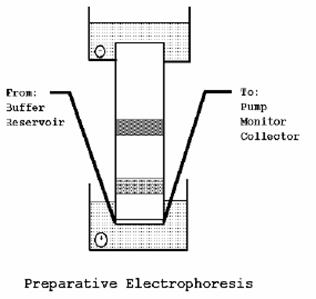
圖6、preparative electrophoresis儀器簡圖:在膠體底部放置透析膜,
當蛋白質跑至底部時會被透析膜收集起來。
a.自由流動電泳(Free Flow Electrophoresis,FFE):
一種以thin buffer film取代傳統固解補充物質(solid support material)(如:gel)的製備式電泳(preparative electrophoresis)方式。Thin buffer film可以是連續或間隔的構造,後者是以兩片之間有空隙的窄玻璃片將thin buffer film內部區隔成多個空室,蛋白質樣本可在電泳進行時連續注入空室中。而此電泳方式主要是依據帶電粒子在電泳中的移動位置(electrophoretic mobilities,EPMs)或等位點(isoelectric points,pIs)將其分離。所能分離粒子的大小範圍小至分子大至細胞皆可。
電流方向是和電極與sample流動方向相垂直。可在sample仍在
流動時加入電流(continuous FFE),或在sample流動的短暫停滯期間
施加(interval FFE)。
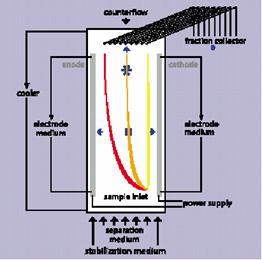
圖7、FFE( Free flow electrophoresis )結構簡圖:蛋白質的樣本利用電場而分開在不同
的pH梯度上,最後收集在不同的空室中(6)。

圖8、FFE儀器的外觀:ProTeam FFE(6)。
b.等電點分離器(Rotofor Cell):
The BioRad Rotofor Cell System 是利用蛋白質的等電點來分離蛋白質,內部具有一個圓柱形的集中的空室,另分隔為20個隔間(compartment)。蛋白質樣本以內含水及兩性電解質(ampholytes)的混合物(若含特殊蛋白質而需增加其溶解度時,也可包含中性的清潔劑)稀釋後,即可注入該集中空室中。一般建議提供的能量為12-15W,電泳進行時間約4小時,電壓隨電泳進行而增加,直到蛋白質依pI分離至二十個不同的小空室中。此儀器的兩大特色為:
*保持待測蛋白質的生物活性。
*能處理大量的total protein。
因此對於分離low abundant protein而言,為一種不錯的初步分離方式。


圖9、BioRed Rotofor Cell System:左圖為集中空室(focusing chamber)的外觀,
又突圍等電分離器的全觀,包含集中空室及二十個小空室。
1、各種蛋白質分離技術
平板凝膠電泳(Slab-gel electrophoresis)是一種蛋白質分離技術,其能同時分析一維的形式(1-D format)與解析大量的蛋白質在二維的形式(2-D format)情況。除此之外,對於需要特殊之儀器才能達到蛋白質分離的技術-高效液相層分析HPLC(Hight performance liquid chromatography)、毛細管電泳CE(Capillary electrophoresis) -而言,平板凝膠電泳相對上較符合經濟效益。然而,平板凝膠電泳之成本較低,但是它在分離上有需多的限制,例如:對於疏水性與膜蛋白之溶解度限制大、受限的動力範圍(dynamic range)、對於鹼性或酸性蛋白質難以有效分離、低的敏感度與難以定量、無法自動操作等。另一方面,高效液相層分析、毛細管電泳在分離技術的限制上與平板凝膠電泳截然不同。
|
Function |
CE |
SGE( Slab-gel electrophoresis) |
HPLC |
|
Automation speed |
Yes |
No |
Yes |
|
Sensitivity |
nm-fematomole |
Slow,minutes-hours |
Minutes |
|
Sample size |
nL |
μM |
μM |
|
detcetion |
On-column |
Staning,fluorscence |
Off-column |
|
Quantitation |
Yes,area under peak |
Possible,but not simple |
Area under peak |
|
Multi samples |
Yes,96-384 capillaries |
Yes |
No |
|
Multidimensional |
Yes |
Yes |
Yes |
表 1:各種蛋白質的分離技術在功能上的比較(7)。
2、如何分離蛋白質
在此我們必須確定欲要分離的蛋白質是哪一種類型,大致有:來自於混合物(cell lysate,cell culture susperntant,serum)中的單一特蛋白質、一群蛋白質或是一個細胞或組織中所有的蛋白質。因此就不同來源的蛋白質,有不同的分離技術,以及各技術間的結合而形成多維之方式(multidimensional approach)來分離蛋白質,或是選擇何種分離的特性 來分離,例如:親和力、等電點(Isoelectrophoresis focusing)、離子交換(Ion exchange)、分子排除(Size Exclusion)就顯的重要。沒有單一的色層分析(chromatography)或是電泳(electrophoresis)能完成具有複雜性較高的蛋白質之分離,因此必須將蛋白質分解成數個peptides,再分離會比較有利益,且能簡化蛋白質於定性上的難度而能得到準確的結果。
3、不同蛋白質的分離
(1)特定蛋白質的分離(Selective separation of a specific proteins)
利用親和性HPLC或CE(affinity HPLC or affinity CE)來達成一特殊的蛋白質分離。而親合力的原理是以具有生物活性的物質結合至具專一與可逆性之欲要被分離的分子來完成。生物活性的物質有:抗體(antibody)、(金屬)metals、醣結合蛋白(lectin)、concanvalinA、 生物素(biotin),適合體(aptamers)、RNA or DNA fragment等。至於特殊的蛋白質在分離上,HPLC方面其是以裝有原料的管柱(column)之活性表面來吸引蛋白質分子而達到分離的目的,而CE則是以管柱表面具活性的部分來吸引蛋白質分子而達到分離的目的。來自於蛋白質混合物(protein mixture)、血清、細胞溶解物(cell lysate)等欲分離樣品,被注入至管柱,其後感興趣的蛋白質分子被其親和物質(affinant)抓住而其它的的化合物則經由管柱流出。圖十是以antibody來做為生物活性物質而達成生物親和性之色層分析(bioaffinity chromatography),而在感興趣的蛋白質分被親和物抓住後藉著改變緩衝液的性質、pH、離子強度、溫度而將感興趣的蛋白質洗出。


圖10:利用抗體的親合力抓特定的蛋白質(8)。
(2)、分離一群蛋白質(selective separation of a group of proteins)
能利用上述利之親和性HPLC或CE(affinity HPLC or affinity CE)來達成蛋白質分離之外,還能逐步依照感興趣的蛋白質的性質(例如:疏水性、電荷、極性、大小與鍵結特性)來改變實驗情況。這些改變有(1)移動相的的性質,例如:梯度的斜率(slope of the gradient)、有機修飾物的百分率(% organic modifier)、緩衝液的pH、鹽類的濃度、ampholite’s pH range。(2)管柱的形式,例如:RP(C-18),離子交換(ion exchange)、大小排除(size exclusion)、膠體過濾(gel filled)、常態相(矽石)﹝normal phase(silica)﹞、親和性(抗體的種類、lectin、aptimers、metals、DNA等)。因此實驗的設計通常依賴感興趣蛋白質的疏水性、電荷、等電點或親和性。例如:含有組胺酸的肽類(histidine-containing peptides) 對於銅有金屬親和性;又含有磷酸鹽的肽類(phosphopeptides)對於鎵有金屬親和性;Lectin則在對醣蛋(glycoprotein)有高度親和性。
(3)、分離細胞或組織中所有的蛋白質(selective separation of all proteins in a cell or tissue)
必須以多維(multidimensional)的方式來分離細胞或組織內的所有蛋白質,即利用平板凝膠電泳、毛細管電泳、高效液相層分析多個方法來達成分離;或是利用同一個分離方式中不同的分離機制,例如:分割(partition)、吸附作用(adsorption)、親和性、大小排除(size exclusion) 來達成分離。以2-D電泳分析 (2-D PAGE)為例子,在分離蛋白質分子時先以蛋白質的電荷做為基準(第一維),之後再以蛋白質分子量大小來更進一步分離蛋白質分子(第二維)。除了2-D gel electrophoresis之外,其它的多維的方式(multidimensional approaches)而使蛋白分離之方法,例如分析者能結合二種不同的HPLC(或CE)分離機制或結合平板凝膠電泳、毛細管電泳、高效液相層分析模式來分離蛋白質。分析者也能利用Reversed-phase HPLC之後緊接著HPLC或是CE之後緊接著Reversed-phase HPLC或是CZE之後緊接著IEF- CE。
|
SGE |
HPLC modes |
CEC mode |
CE mode |
|
Size |
HIC Reversed-phase |
HIC Reversed-phase
|
CZE |
|
Charge |
Ion exchange
|
Ion exchange
|
IEF |
|
IEF |
Size exclusion Affinity Normal phase |
Normal phase |
SDS-PAGE Affinity MEKC
|
表.2:各種不同分離機制相互結合而完成蛋白質分離的目的。
4、分離蛋白質的多維方法(Multidimensional separations of proteins)
(1)、二維電泳分析(2-D gel electrophoresis)
分離大量蛋白質時,我們通常採用2-D gel electrophoresis(研究蛋白質體學的必要技術),但在最近的研究中發現2-D PAGE和MS一同作用時,只能偵測出量大的蛋白質,但即使有缺陷,此技術仍是我們最廣為使用的細胞或組織中蛋白質多維分離技術。從許多的實驗室及疾病研究經驗中也告訴我們2-D gel electrophoresis 和2-D maps比較起來是一個相當可靠的方法。
蛋白質被2-D gel electrophoresis分離染色後,gel上會有點顯露出來,加入trypsin將蛋白質切成一段段的peptide fragments,MS會將一個個的fragment定量,我們就能和DNA或蛋白質資料庫中的預測做比對,比較peptide fragments的分子量,以確定這是什麼蛋白質。
蛋白質體學目前受到2-D gels解析能力限制,如果我們用spot detection在標準的2-D gel format中可以看到3000-10000個proteins(即點),但這些點代表的是在每一細胞蛋白質族群中超過10000個分子的蛋白質,至於其他的偵測方法,也無法偵測低於每一細胞1000個分子的蛋白質,ex.Fluorescence,imidazole zinc,or silver staining。假設每一細胞有109個蛋白質分子和50000-500000個mRNA分子,一個mRNA又可以製造出許多蛋白質,我們暫定一個哺乳動物的gene平均在gel上產生五個點,每一細胞10000個active gene,就可以在gel上產生50000個點,所以我們在2-D gel上看到的3000-10000個點只不過是7-24%出現最豐富的蛋白質罷了。因此依此理論,76%的蛋白質都低於偵測限制,且大部分在每一細胞中不超過10000個分子。
(2)、高效液相層分析和毛細管電泳共用的多維分析( Multidemensional HPLC,CE and HPLC-CE protein/peptide separations)
分離細胞或組織中的所有蛋白質並不是一件簡單的事情,人類細胞中有數以千計的蛋白質,經代謝後更會產生大量的peptides,要用單一的色層分析或電泳技術來分離這樣大的一個複合物是不可能的事情,所以我們必須採用多維的方法;在2-D gel electrophoresis中,蛋白質是先被分離,再被trypsin代謝,最後peptide片段才被MS分析出來。但在色層分析和毛細管電泳的多維方法中,蛋白質是先經酵素作用然後才被分離,這麼做的好處是月生肽片段和完整的蛋白質比起來溶解度較高也較易分離,尤其是對厭水的膜蛋白而言,壞處就是我們必須解決數量更大的peptides。
5、利用高效液相層分析和毛細管電泳可增加蛋白質的濃度(On-column HPLC and CE protein concentration)
高敏度對蛋白質體學的研究是很重要且決定性的一點,HPLC和CE的優點是它們可以把稀薄的樣品在分離前於管柱上濃縮,在HPLC稀薄溶液藉由動向的巧妙操作可以在分離前被濃縮於管住的頂端,CE則採取不同的濃縮方法,包括antibodys的運用、接上C-18、eletrophoretic techniques(ex.stacking)、IEF或isotachophoresis(ITP)。事實上蛋白質混合物經愈多次處理損失愈多,CE適時提供一項優點,分析者可將細胞引入毛細管或微流管道,溶解細胞再分離蛋白質,所以我們可以除去管外操作時蛋白質流失的缺點。
6、偵測
不像2-D gel electrophoresis在偵測、定量已分解蛋白質還需要染色或其他的步驟,HPLC、CEC、CE技術中蛋白質和peptides可直接用UV absorption、fluorescence、MS on-line偵測,peptide bond CO-NH在185-220-nm波長有很強的吸光力,讓我們偵測出蛋白質的存在,偵測敏感度在波長區間的底端增加,然而分析者應該小心選擇不會吸收特定偵測波長的buffer。蛋白質和peptides結構中含有aromatic amino acid residue者可被UV absorption at 275-280-nm和fluorescence in the 200-300-nm波長區間偵測出,但用UV absorption偵測敏感度較高。
7、蛋白質的定量分析(Proteome quantitation strategies)
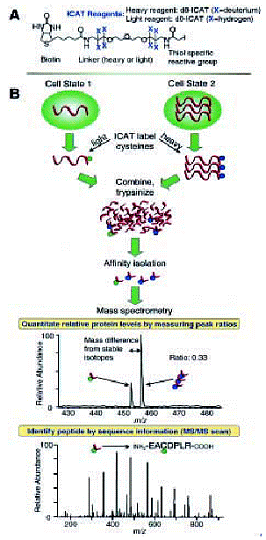 細胞或組織中蛋白質的定量並不是一件簡單的事情,過去我們用2-D
PAGE測量蛋白質在表現階段時的變化(藉由比較點的明暗度),但如之前提到的,2-D
gel這個技術缺乏sensitivity、reproducibility和quantitative
precision,所以現今又發表了革新的技術來改善─ICAT(Isotope
code affinity tags),如圖11所示。
細胞或組織中蛋白質的定量並不是一件簡單的事情,過去我們用2-D
PAGE測量蛋白質在表現階段時的變化(藉由比較點的明暗度),但如之前提到的,2-D
gel這個技術缺乏sensitivity、reproducibility和quantitative
precision,所以現今又發表了革新的技術來改善─ICAT(Isotope
code affinity tags),如圖11所示。

8、蛋白質的鑑定(Protein identification)
Gel electrophoresis通常用來做蛋白質的純化與初級分析,未知蛋白質被gel electrophoresis分離後用MALDI-TOF MS做peptide mapping以快速鑑定,鑑定的方法通常都是拿被酶切斷的蛋白質即peptide fragments和sequence database中的每一個蛋白質peptide fragments比對質量,通常都要得到好幾個peaks才能確定它到底是哪一個蛋白質,有時候我們也會得到多於一個的解,據此我們自然就要用MS/MS得到的另外訊息來縮小搜尋,然而這需要更複雜更花時間的樣品分析。雖然ESI-MS/MS比MALDI-TOF的信賴度高,但MS還是有它的短處在鑑別有相同分子量的amino acids如Q與K或E與N而導致peptide sequence有不正確的排列順序。因此發展出了新的方法來解決此問題,其一用CE來做peptide mapping,此方法中peptide的遷移率依理論模型(以peptide width、charge、residue mass為基礎)做預測,另外的方法就是Nanoelectrospray CE-MS,以electrophoretic mobility-assisted identification of proteins的原理達到,這個方法依據Offord發展出的理論模型,說明peptide的電泳遷移率和它的分子量與帶電量有關,可以作為一個功能的選擇,帶電量上的差可以用來認出MS不能分辨的amino acid,如Q與K,而找出正確的peptide sequence辨認出該蛋白質。
9、結論
蛋白質體學的研究技術日新月異,不同技術已在科學文獻中被出版。加上自動化的技術,更能在電泳分離後的peptides連線至MS來做定量與該蛋白質序列分析。而蛋白質體學的持續研究,也將對製藥業、臨床化學、毒性物質的辨識等,將有重大的貢獻。由於Human Genome Project之故,使Genome的研究工作達巔峰,然而在細胞中主要執行各項生理機能、代謝的生命分子是-蛋白質。Proteome = PROTEins expressed by the genOME.由此知gene的產物是protein,代理了gene從各種生化反應,因此蛋白質體學對近代分子層級的生物學研究扮演重要的角色,然而由於蛋白質體不像基因體來的單純,因此研究工作更加複雜,加上現在無任何技術能將感興趣之蛋白質大量複製、放大(gene能利用PCR來放大感興趣的DNA sequence),所以在非常微量的sample中進行分析著實不易。
新的儀器及分離技術不斷的改進,使得對蛋白質分子檢測時的敏感度(sensitivity)大幅度增加,但相對上較不符合經濟效益,因為儀器相當昂貴。而藥物的開發是可說是蛋白質體研究工作的運用,對於各種疾病將能獲得新的藥物治療,這也許是生命科學持續進步帶來劃時代的改變。
1. http://www.ncbi.nlm.nih.gov/books/bv.fcgi?call=bv.View..ShowSection&rid=mcb.figgrp.d1e1
2. http://www.ncbi.nlm.nih.gov/books/bv.fcgi?call=bv.View..Show
3. http://ac220.ac.ntu.edu.tw/Protein/Purification/P1.htm#鹽析
4. 中央研究院 生物化學研究所 吳啟裕博士提供
5. Amna Butt. Matthew D. Davison , Gaskell , Stephen G. Oliver , Robert J. Beynon , 2001 , Chromatographic separations as a prelude to two-dimensional electrophoresis in proteomics analysis. Proteomic. 42-53
6. http://www.tecan.com/index/com-pr-ap_so-entry/com-pr-ap-entry-proteomics/com-pr-ap-pr-fractionation/com-pr-ap-prot-ffe.htm
7. Electrophoresis 2001,22,3630,Table 1
8. Electrophoresis 2001,22,3631,figure 1
9. Electrophoresis 2001,22,3632,Table2
10. Electrophoresis 2001,22,3636,figure2