蛋白質分析
口述:莊榮輝 老師
整理:楊晴惠、張靜農、呂元馨
(d) Bradford Method(Comassie Brilliant Blue G-250)
(e) Special Binding Groups(heme)
5. 電泳膠體蛋白質的溶離:使用little blue tank
1. 組合不同數目胺基與酸基可合成多種 pI 的混合物:商業上之應用
現階段的酵素活性分析方法
1. 蛋白質定量-利用蛋白質的分子構造
2. 酵素活性分析-利用有催化活性的巨分子
3. 膠體電泳法-利用Native-PAGE及SDS PAGE
4. 免疫轉印法-利用抗體的專一性
5. Proteomics-利用解開的基因序列加上大量database
1. 各種蛋白質定量法:(圖1)
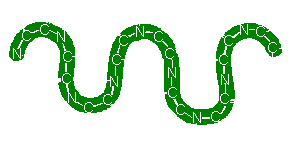
蛋白質的主要架構:蛋白質分為脊股和枝鏈(side chain)兩部分,脊股是以NCCNCCNCC為主要的結構,在C和N之間為月生肽鍵(peptide bond),加上一些帶有極性或非極性的蛋白質枝鏈所組成。
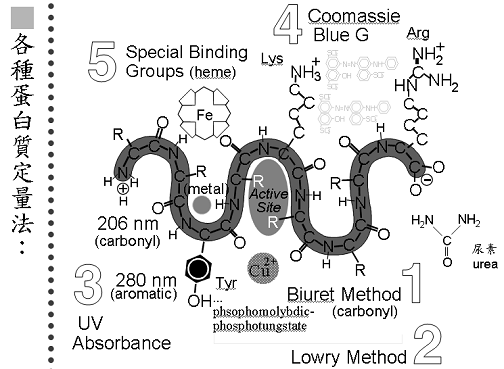
圖1:蛋白質定量方法—1.Biuret Method;2.Lowry Method;3.UV Absorbance;4Comassie Blue G;5.Special Binding Groups(heme)
利用蛋白質上的兩個carboxyl group 接上Cu2+離子,當蛋白質接上銅離子之後,會使得銅離子變色,呈現紅色,因此此種方法是利用蛋白質上的脊股來做定量,但是因為呈現的強度不強,所以有更多的定量發法出現,但是此種方法屬於很準確的方法,因為脊股上衣定有carboxyl group所以一定可以測出來只是非常不精確。
為何此種方法稱為Biuret Method?是因為銅離子接上的地方很像兩個urea(尿素)因此稱此種方法為Biuret Method。
此法分為兩個步驟,首先使待測物與鹼性銅離子作用,由於蛋白質中的peptidic bond會鹼性銅離子作用產生藍色,而蛋白質中的Tyrosine及tryptophan與Folin-Ciocalteu(phenol)試劑作用產生更深的藍綠色,此在波長660nm有很強的吸光,故測定OD660,並與已知濃度蛋白質比較,換算出待測液中蛋白質含量。一般多以BSA為標準蛋白質(standard protein)。
分為兩種(1)UV 206nm:利用蛋白質脊股上的carboxyl group在波長206 nm有吸光的特性來測量。(2)UV 280nm:利用蛋白質上的苯環(aromatic group),因為苯環有共振性,在 波長280nm有吸光性,利用此特性來定蛋白質的量。利用OD206來定蛋白質的量較準確也較精確,但是因為UV 206nm波長較短非常的難做,所以多改做280nm 的吸光值,而且OD206容易受空氣中的CO2和O2干擾,所以即使OD280較不準確仍多使用此方法做蛋白質的定量分析。
(d) Bradford Method(Comassie Brilliant Blue G-250)
原理(圖2):Comassie Brilliant Blue G-250(在此簡稱CBG)在酸性環境下為茶色,當蛋白質加入後,蛋白質會與CBG結合,使CBG轉變為藍色,當愈多蛋白質和CBG染劑結合則表現的藍色就愈深,所以蛋白質的量和的顏色深淺呈現正比的關係。
不同的蛋白質和結合的能力會有所不同所以在蛋白質的定量時必須再和一組已知量的蛋白質而做出來的標準曲線來做比較(通常是將待測量的蛋白質的吸光值帶入標準曲線的直線方程式中以求得相對的量),做標準曲線的蛋白質通常是BSA、IgG,這些做標準的蛋白質,在做標準曲線時,每個不同量的蛋白質都要是相同種類的蛋白質,不可以BSA或IgG混合在一起做標準曲線。
Coomassie Brilliant Blue G-250
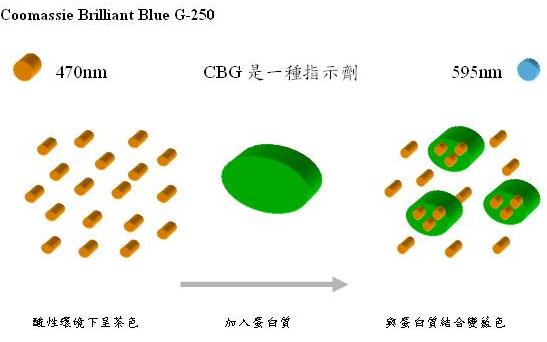
圖2: 利用Comassie Brilliant Blue來定蛋白質的量,當呈現的顏色愈深即表示所含的蛋白質含量高。
(e) Special Binding Groups(heme)
利用蛋白質上可以接上某些metal group來定蛋白質的量。
(a)在所以的蛋白質定量方法中以波長 206nm的吸光度來測試最精確也是最準確的方法,但是因為此種方法較難測,所以先在多採用Bradford Method的方法,因為此種方法為一種折中的方法,為其他方法中較為不錯的方法。
(b)Biuret Method的準確度較高,但是靈敏度不高而且精確度不高,其他的特性為呈色快速、有腐蝕性、銨離子干擾大。
(c)Lowry Method的精確度高,但是準確度不高,其他的特性為呈色較慢、有多種離子會干擾。
(d)Absorance 280nm的準確度和精確度都不高,但是因為方便所以仍較為常用。
(e)Absorance 206nm的精確度和準確度都高,但是因為亦受干擾較不常用。
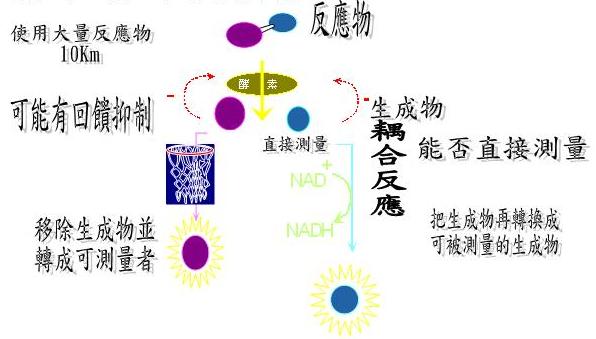
1. 酵素反應及偵測方法(圖3)
使用大量的反應物(10×Km)->加入酵素->測生成物
測生成物的方法可分為直接測量和間接測量:
直接測量-所得的生成物可見,而可以直接偵測。
間接測量-所得的生成物不能直接測量,利用耦合反應(coupling reaction),在反應後加上dehydronase使酒精的H+轉移到輔脢NAD+,使得NAD+變為NADH,酒精變為乙醛,因為NADH在OD340的吸光值會改變,所以當生成物愈多,NADH的吸收值會高,利用此種方法可以偵測生成物的量。

遵守SEPt的原則
S:基質要過量(10×Km)
E:酵素量要適中
P:生成物要能測得
t:反應時間要恰當
另外兩個原則為1、buffer要確定。2、溫度要確定。
(a)正確的反應時間為反應曲線的斜率,並非直接將生成物除於所需的時間,因為在每個時間生成物不一樣,這樣算起來生成的速率也會不一樣,所以必須求曲線的斜率來作反應的生成速率(即酵素活性)。
(b)因此反應時間必須適當,否則時間太短,會找不到生成物的生成,也不能算其斜率。
(c)若反應曲線為直線上升,求不到其斜率,須算一部份的面積,求切線當作生成速率。
(a)濾紙電泳cellouse-蛋白質分子量過大,不易隨著液體而跑,而且易造成蛋白質變性。
(b)薄層電泳(TLE)cellouse acetate-將濾紙磨碎做成膜狀,但仍會造成蛋白質的變性。
(c)膠體電泳starch gel-利用多醣類來做電泳,但是因為不好做所以現在改為化學合成。
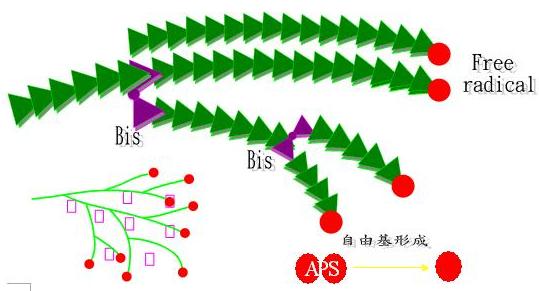
2. 膠體的聚合反應(圖4)
=>利用Ammonium persulfate(Free redical initatior)
當單體acrylamide(monomer)加入Ammonium persulfate,因為Ammonium persulfate 榮於水中會形成自由基即(O4S-SO4)2-->2SO4-,此自由基可催化acrylamide聚合形成長鍊的分子結構,在聚合反應中會加入Bis(acrylamide)架橋使聚合產生分枝,並有TEMED作為催化劑,催化acrylamide及Bis-acrylamide的聚合。
※
 蛋白質是在acrylamide孔洞中移動,acrylamide濃度愈高孔洞愈小,acrylamide濃度愈低孔洞愈大,所以當要分離小蛋白質分子時要將acrylamide的濃度弄得較高一點。
蛋白質是在acrylamide孔洞中移動,acrylamide濃度愈高孔洞愈小,acrylamide濃度愈低孔洞愈大,所以當要分離小蛋白質分子時要將acrylamide的濃度弄得較高一點。
3. 電泳膠體系統的組成(表1)
依膠體的緩衝液pH值不同可以分為焦集膠體(stacking gel)及分離膠體(separating gel or稱 running gel),其pH值分別為6.9及8.3,焦集膠體的目的可以使蛋白質全部聚集起來,從相同的起跑點開始移動,因為此層的緩衝液包含氯離子及glycine,因為氯離子分子量較小跑得較快,glycine分子在焦集層因其pI值和環境pH值接近所以不帶電而造成跑得較慢,所以在氯離子和glycine之間有一段缺乏帶電分子或離子移動的空間,電壓較高,會促使帶負電的蛋白質快速往正極移動,等到全部都堆積在焦集層形成一薄層,如同在同一個起跑位置上一樣,glycine進入pH為8.3的separating gel後姐離成帶負電的分子,開始向正極移動,其他的蛋白質分子也開始移動,依其分子量或pI值來分離蛋白質。
|
電泳系統 |
緩衝液 |
pH |
膠體濃度 |
|
1上層(負極)緩衝液 |
Tris-glycine |
8.3
|
|
|
2樣本溶液 |
Tris-glycine |
8.3
|
|
|
3焦集膠體 |
Tris-HCl |
6.9 |
5﹪
|
|
4分離膠體 |
Tris-HCl |
8.3 |
5-20﹪
|
|
5下層(正極)緩衝液 |
Tris-glycine |
8.3
|
|
表1: Discontinuous-PAGE的主要組成,主要在焦集膠體和分離膠體緩衝液所造成的pH值不同而稱為dis-PAGE。
原始的電泳膠體為disc-PAGE(native-PAGE),在分離蛋白質時需要考慮其分子量及pI值,比較不方便,所以現在的電泳膠體會在膠體中加入SDS(sodium dodecyl sulfate),SDS為一介面活性劑,分為極性頭部及非極性尾巴,非極性尾巴會深入蛋白質的核心部分,而極性頭部會和水接觸,並會使蛋白質被denature,變成一線狀分子在膠體中行動,且因SDS會在蛋白質分子表面均勻覆上一層負電,所以蛋白質多帶負電(圖 5),因此蛋白質在膠體中行動不需考慮其帶電性,只要考慮其分子量,這樣一來分離蛋白質就容易多了。

![]()
![]()
![]()

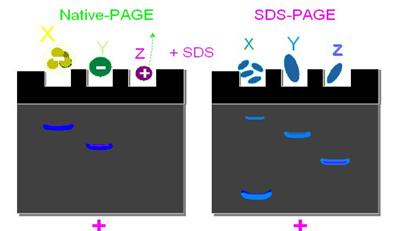
例如有三個蛋白質(圖6):protein X 、proteinY、proteinZ在不同的PAGE中會表現出不同的情況。


 X
Y
Z
X
Y
Z
![]()
圖6: 三種不同性狀的蛋白質:蛋白質X由四個單體形成;蛋白質Y、Z皆由一個單體形成。
在native-PAGE中因為proteinX和proteinY的pI值都低於pH8.3所以都帶負電,但是proteinZ的pI值高於pH8.3所以帶正電,因此proteinX和proteinY會往正極跑,但是proteinZ並不會,而會往相反的方向跑,所以只會得到proteinX和proteinY,因為proteinY的分子量大於proteinX,所以跑得較慢,最後的結果為proteinX跑得最快。(圖7-a)
在SDS-PAGE中,因為蛋白質全部都帶負電,所以只要考慮分子量,而且SDS會使蛋白質變性,所以proteinX會denature形成monomer,所以最後為proteinX跑的最快,次之為proteinZ,最後才為proteinY(圖7-b),如此一來,可完全依照分子量去區分蛋白質大小。(表2)
|
Protein |
Quaternary Structure |
Molecular Weight |
pI |
Mobility
Native -PAGE SDS -PAGE |
|
|
X |
Tetramer |
(40000)×4 |
5.8 |
Slow |
Fast |
|
Y |
monomer |
88000 |
5.2 |
Fast |
Slow |
|
Z |
monomer |
66000 |
9.3 |
Upward |
Medium |
表2: 蛋白質X、Y、Z在Native –PAGE及SDS –PAGE中泳動的快慢
(a) (b)
圖七 (a) Native-PAGE (b) SDS-PAGE
X為tetramer (分子量:40000×4),Y(分子量:88000)、Z(分子量:60000)為monomer;pH8.3之情況下:NATIVE PAGE ( X,Y 帶負電;Z帶正電 );SDS-PAGE ( 皆帶負電 )
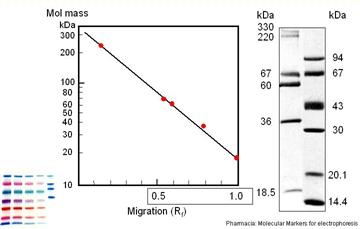
圖8:單元體分子量的測定(SDS-PAGE),由電泳結果之樣本移動距離依內插法作一線性圖求分子量。X軸:分子量(kDa) Y軸:泳動率(Rf)
根據結構之特徵(圖9),即因含有特定side chain而和染劑產生作用,故達到染色效果,有兩種方法,第一種方法:將硝酸銀溶於硝酸溶液中,產生銀氨錯離子,用此可染蛋白質中含有Lys部分,銀離子會還原成金屬銀而附著在Lys之side chain上,產生深褐色反應產物。第二種方法:使用coomassie blue R (red),調整其pH值,使其呈藍色,可染Lys及Arg之side chain部分,而達到染色效果。
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
圖9: 兩種主要蛋白質染色法:1. Ammoniacal silver 2. Coomassie blue R
5. 電泳膠體蛋白質的溶離:使用little blue tank
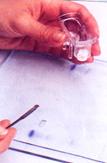
直接挖出膠體進行溶離或定序(圖10-a),使用little blue tank進行電泳(圖10-b),將溶離出之樣本經通電後會集中在tank一端,有助於吸出並收集(圖10-c)。
![]()
![]()
![]()
![]()


(a) (b) (c)
圖10: (a) 將有色帶呈現之膠體部分挖出並收集在溶離槽內 (b) little blue tank (c) 通電進行電泳前(上)、後(下)之情況,箭頭代表電泳泳動聚集方向



(a) (b) (c)
圖11 :(a) 電泳槽(左)、鑄膠器(右) (b) 轉印三明治(左)、轉印槽(中,右) (c) 供電器
一般電泳使用迷你電泳槽,而SDS-PAGE使用其專用之電泳槽
![]()
通電使膠體上之色帶轉印至nitrocellulose上(圖12-a),先以ponceau染nitrocellulose,確定轉印是否成功,有紅色色帶呈現即表示轉印成功,在以清水洗淨,進而已抗體偵測呈色(圖12-b)。
![]()
![]()
![]()
![]()
![]()


![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
![]()
![]()



![]()
![]()


![]()
利用抗原抗體結合之原理,原本使用放射線標定於抗體上,後來改為使用酵素,先由一級抗體和轉印紙上的特定抗原結合,此抗原即是要找之peptide,一級抗體可自行製備,通常選擇異源性且分子量上萬之蛋白質去引發抗體產生而可作為一級抗體,再使用抗一級抗體之二級抗體去和一級抗體結合,其上已標定好酵素,但若先標定一biotin則可達到放大信息的效果,因biotin和streptavidin有很強之結合力,其解離常數為10-20,而streptavidin又能再和另三個biotin結合,此三個biotin尚在各自接一酵素,可放大信息三倍,有時二級抗體上會標定金屬,如:金(圖13-b),抗體抗原之專一性結合完畢後,加入受質作用,受酵素催化形成不溶性產物而堆積在酵素作用周圍,就能顯示欲找之樣本所在(圖13-c)。免疫轉印法之靈敏度比硝酸銀染色法高。
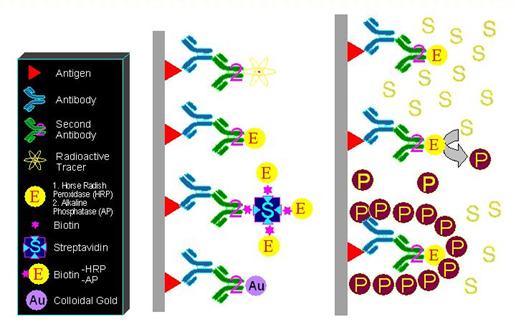
(a)
(b)
(c)
圖13:免疫轉印之(a)選用專一性組合單元,及(b)種類,與(c)呈色機制
等電焦集法
蛋白質之帶電性會隨著環境pH而改變,當處於等電點時,其凈電荷為零,故在跑電泳時會停止移動,利用此原理能進行等電焦集法進而達到分離之目的(圖14)。

圖14:等電焦集的作用機制-pI值為7之蛋白質進行等電焦集電泳,當泳動至pH值為7之環境時會停止移動蛋白質在低於其pI值之環境下,其淨電荷為正;若在高於其pI值之環境下其淨電荷為負。
1. 組合不同數目胺基與酸基可合成多種 pI 的混合物:商業上之應用

 藉由控制分子上正負電性基團之數目而造成多種pI之混合物。第一次元先進行等電焦集電泳於一長條平面上,接著再進行第二次元之SDS-PAGE電泳,即先將樣本以等電點不同之特性先分離,再進行和等電焦集電泳方向垂直之SDS-PAGE電泳,利用分子大小不同作區分(圖15)。
藉由控制分子上正負電性基團之數目而造成多種pI之混合物。第一次元先進行等電焦集電泳於一長條平面上,接著再進行第二次元之SDS-PAGE電泳,即先將樣本以等電點不同之特性先分離,再進行和等電焦集電泳方向垂直之SDS-PAGE電泳,利用分子大小不同作區分(圖15)。
將樣本進行2D電泳,從膠體中挖出欲分析之部分,將其中之蛋白質溶離並用酵素切成小片段,接著將這些小片段分兩路進行不同分析,一方面進行MALDI-TOF質譜儀分析得各小片段之分子量;另一方面以HPLC依極性不同進行分離,將小片段隻胺基酸進行由N-端至C-端之定序,最後將所得之小片段資料,包括分子量及序列,進入現有蛋白質資量庫進行比對,最終目的是要求出是什麼蛋白質(圖16)。
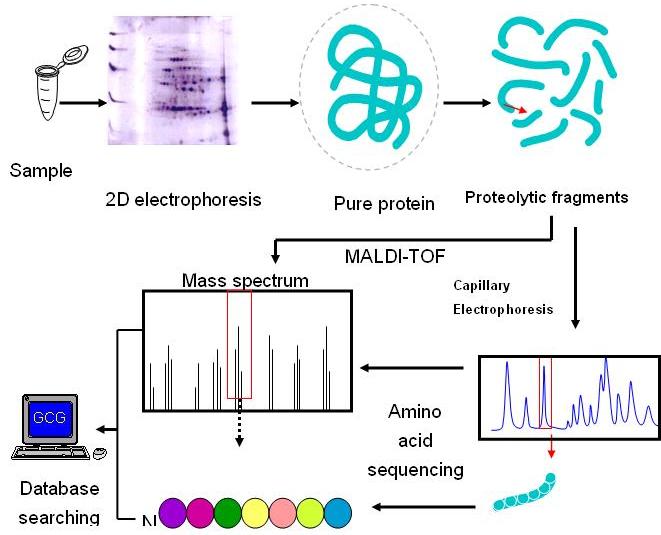
![]()
參考資料
1. http://140.112.78.220/~juang/ECX/index.htm.